胰导管腺癌(PDAC)起源于胰腺上皮。尽管发病率低,但它是最致命的癌症类型。尽管预后不良很大程度上是继发于被诊断为晚期疾病的患者的高比例,但PDAC的预后也受到这种恶性肿瘤固有的生物侵略性和高转移潜力的影响。治疗方案仍然有限,过去几十年来进展甚微。在同时接受化疗和/或放疗的患者中,手术结果会有一些改善,然而,由于PDAC对所有治疗方案的强烈耐药性,对长期生存的影响微乎其微。因此,迫切需要1)更好地了解PDAC的生物学特性;2)制定早期发现和预防方案;3)识别新的治疗策略,以提高生活质量和生存率。在这篇综述中,首先,我们将总结PDAC发病机制的知识现状,特别是对引起治疗耐药的分子特征的关注。然后,我们将简要回顾当前和新兴的方法在PDAC护理。 Lastly, we will highlight the integrative approaches in the light of new experimental and clinical research conducted with the aim of moving towards personalised therapy in patients with PDAC.
胰导管腺癌,个体化医疗,综合肿瘤学
PDAC是最致命的癌症之一,其发病率与死亡率相等[1,2]。PDAC是世界上第12种最常见的癌症,也是第7种最常见的癌症死亡原因[3,4]。PDAC的发病率和死亡率在西方国家最高[4]。在过去20年里,无论男女,其发病率都在稳步上升[3,5]。这种疾病前景黯淡的主要原因是大多数(约80%)患者被诊断为晚期疾病,严重的恶病质和不良的代谢状态迅速导致发病率和死亡率[6]。目前,尚无有效的筛查和早期检测方法可在恶性前期诊断PDAC。虽然手术切除胰腺肿瘤提供了最好的生存机会,但只有少数(10- 15%)患者在诊断为[6]时可以接受根治性手术。即使在手术后,5年生存率也低于4%,主要原因是微转移已经确定,最终导致局部和/或全身复发[3]。在同时接受化疗和/或放疗的患者中,手术结果可能会有一些改善,然而,由于该疾病对当前治疗方案的强烈耐药性,对长期生存的影响微乎其微。因此,迫切需要新的临床管理策略。
在这篇综述中,首先,我们将总结PDAC发病机制的知识现状,特别是对引起治疗耐药的分子特征的关注。然后,我们将简要回顾当前和新兴的方法在PDAC护理。最后,我们将根据新的实验和临床研究强调综合方法,旨在向PDAC患者的个体化治疗迈进。
胰腺是一个细长的小叶腹部器官,包含血管和淋巴管,神经和排泄管。它可以分为四个部分:头,脖子,身体和尾巴(图1a)。靠近十二指肠的胰腺较宽的一端称为头,中间的部分称为体,其余的部分称为尾,一直延伸到脾门。固定于腹膜后,与十二指肠、胆总管、胃、横结肠、左肾、左肾上腺和脾脏密切相关,这意味着侵袭可在多个方向发生(图1a)。
胰腺是消化系统和内分泌系统的双重器官。内分泌胰腺通过分泌激素调节代谢和葡萄糖稳态。它由称为朗格汉斯岛的细胞群组成,按分泌功能分类:β细胞产生胰岛素,α细胞产生胰高血糖素,δ细胞产生生长抑素,PP细胞产生胰腺多肽激素(图1b)。外分泌胰腺由腺泡细胞、导管细胞和中心腺泡细胞组成(图1c)。当腺泡细胞合成消化酶进入酶原颗粒时,导管细胞产生富含碳酸氢钠离子(NaHCO)的碱性液体3.)和粘液将酶原冲进肠道[7]。在生理条件下,NaHCO3.分泌中和腺泡细胞分泌的酸性物质,防止消化酶在腔内聚集,中和从胃进入十二指肠的酸性食糜。然而,在病理生理条件下,强化和延长酸化可通过沉淀蛋白质和/或粘稠液和破坏细胞间连接引起导管管腔阻塞,最终可能导致胰腺炎,这是众所周知的PDAC的危险因素[7,8]。胰腺的内分泌和外分泌功能由神经输入和激素机制的综合系统调节。

图1.一个)胰腺的大体解剖及其在腹腔中的位置。的说明b)朗格汉斯岛和c)胰管系统。
虽然PDAC的确切病因尚不清楚,但流行病学研究已经确定了不可改变(遗传)和可改变(非遗传)因素都是导致疾病发展的因素(图2)。最新的监测、流行病学和最终结果项目(SEER)和国家卫生统计中心的数据表明,约1.5%的男性和女性在一生中[9]会被诊断为PDAC。但目前对于此类高危患者的评估、筛查、分层和预防疾病的发生,尚无公认的标准。发病率增加的原因尚不清楚,但肥胖发病率的增加可能是一个促成因素。此外,代谢综合征、糖尿病、老年、吸烟、酗酒、高脂肪饮食、某些微量元素、男性、非“0”型血和非裔美国人种族也是其他危险因素(图2)[10,11]。慢性胰腺炎与PDAC风险增加相关;慢性胰腺炎和PDAC之间的联系在吸烟者或遗传性慢性胰腺炎人群中最强[12]若干研究报告显示慢性乙型肝炎、丙型肝炎、艾滋病和慢性肝炎患者中PDAC风险增加幽门螺杆菌(幽门螺旋杆菌)感染[10,11]。此外,胆囊切除术或部分胃切除术和牙周病史与PDAC风险增加相关[10,11]。大约5-10%的pdac具有家族基础,属于家族性胰腺癌(FPC)[12]的一类。然而,只有少数(约20%)的FPC与已知的遗传综合征或因果基因突变有关(表1)[12]。这意味着PDAC一般不遵循孟德尔遗传,其发展在很大程度上取决于基因和环境的独立和相互作用。在这方面,据报道,吸烟使FPC家庭成员患PDAC的风险增加三倍[12]。
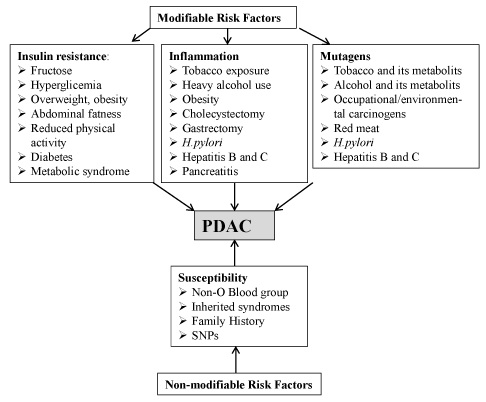
图2.风险因素。(缩写:SNP,单核苷酸多态性;幽门螺杆菌(幽门螺杆菌)
胰腺癌分为两类:外分泌型和内分泌型。超过95%的胰腺癌起源于外分泌胰腺,其中约90%构成PDACs[2]。约65%的PDACs发生在胰腺头部,30%发生在身体和尾部,5%可累及整个胰腺[2]。
PDAC的发展被经典地描述为从导致浸润性癌的前体病变的形态学和分子转化(图3)。遗传学研究已经证实,PDAC可以有前体病变,称为PanIN(胰腺上皮内瘤变)、IPMN(导管内乳头状黏液性肿瘤)和MCN(黏液性囊性肿瘤),起源于胰腺导管上皮[2,13]。然而,最近从小鼠模型和谱系追踪研究中出现的证据也表明,PDAC也可以通过腺泡导管化生过程或通过中心腺泡细胞的膨胀伴随着腺泡细胞[13]的凋亡在中心腺泡-腺泡室中发展。大多数PDACs起源于PanINs(亚PanIN1-3),表现为日益增生和细胞异型性,其特征为极性丧失、核拥挤、核增大、伪分层和深染[2,14]。PDAC以无序的方式生长,浸润胰腺实质,因此肿瘤边缘模糊[2,14]。PDAC的组织学特征之一是一种致密的纤维化基质,称为粘连增生,由细胞外基质(ECM)、间充质细胞、神经细胞、炎症细胞以及血管和淋巴管组成,它们共同构成了肿瘤体积的大部分(高达90%)[15,16]。有趣的是,虽然PDAC在胰腺内部引起强烈的粘连增生反应,但在转移灶[14]中粘连增生可能较弱或不存在。低血管密度是PDAC的另一个特征,阻碍了充足的氧气和营养物质的输送,并导致较大肿瘤[14]的中央坏死。此外,血管和神经周围的侵犯,神经重塑和增强的神经密度和肥大是PDAC[17]的高度特征。淋巴浸润是另一个非常常见的发现,并与淋巴结转移[2]相关。癌细胞可能侵入血管壁或穿透管腔引起血栓形成[2]。 At the time of diagnosis, invasion into adjacent peripancreatic adipose tissue, bile duct, hepatopancreatic duct, and duodenal mucosa is common, causing obstruction of associated duct system [2].
PDAC产生于多种自发和/或遗传突变和表观遗传改变,反映在细胞内信号通路上,这些信号通路通常控制着重要的细胞事件和细胞对外部因素的反应(图3,4)。对刺激亚恶性细胞向PDAC细胞的促进和进展的分子机制的详细了解将最有可能帮助识别新的治疗靶点和药物和化学预防。本节回顾了PDAC中被解除调控的主要分子和信号通路,并与肿瘤和/或间质室相关(图4)。

图3.PDAC从正常上皮向侵袭性生长转移瘤的进展模型。这种进展与高频驱动基因特异性遗传和表观遗传改变的逐步积累有关。这些组织病理学改变伴随着免疫细胞浸润和粘连增生间质反应的增加。(缩写:PRSS1蛋白酶,丝氨酸,1(胰蛋白酶1);SPINK1,丝氨酸肽酶抑制剂,kazal型;MC,肥大细胞;髓源性抑制细胞;TAM,肿瘤相关巨噬细胞;Treg,调节性T细胞;PSC,胰腺星状细胞;ROS,活性氧;基质金属蛋白酶;金属蛋白酶组织抑制剂TIMP;ECM,细胞外基质; EMT, epithelial-mesenchymal transition.

图4.概要模型总结了PDAC的主要特点。图中描述的导致PDAC发展和进展的关键分子途径在文中进行了讨论。缩写:Sig,信号;PancSC,胰腺癌干细胞;PSC,胰腺星状细胞;TGF-β,转化生长因子β;gf、生长因子、IGF、胰岛素样生长因子、INS、胰岛素;EGF,上皮生长因子;HGF,肝细胞生长因子; VEGF, vascular epidermal growth factor; PDGF, platelet-derived growth factor; HH, hedgehog; Cyt, cytokines; MC, mast cell; MDSC, myeloid-derived suppressor cell; TAM, tumour associated macrophage; Treg, regulatory T cell; DC, dendritic cells ROS, reactive oxygen species; MMP, matrix metalloproteinases; SPARC, secreted protein acidic and rich in cysteine; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; IL, interleukin; cyclooxygenase-2, COX-2; CXCL13, chemokine (C-X-C motif) ligand 13; CXCL12, chemokine (C-X-C motif) ligand 12; CXCR2, C-X_C motif chemokine receptor 2, CXCR4, C-X-C motif chemokine receptor 4; NGF, nerve growth factor; mtCFTR, mutant cystic fibrosis transmembrane conductance regulator; Glut, glucose transporter; CA IX, carbonic anhydrase; ATP, adenosine triphosphate; O2,氧气;H+,氢离子;EMT,上皮-间质转化。
PDAC是一种多基因疾病,具有多个高、低种系易感等位基因(表1)。PDAC的遗传异常复杂,有多个染色体的丢失和获得,各种拷贝数改变,微卫星不稳定,以及基因内点突变[18,19]。第一个大型测序研究确定了致癌基因,喀斯特,以及肿瘤抑制基因,CDKN2A,TP53,SMAD4作为PDAC[18]的四个主要“驱动”基因。散发型和家族型和家族型PDAC都有相同的驱动突变喀斯特,CDKN2A,TP53而且SMAD4基因[20]。SMARCA4,背景,EPHA3,FBXW7,表皮生长因子受体,IDH1,NF1基因突变被鉴定为低频驱动因子[18]。最近,对100个pdac的全基因组测序和拷贝数变异分析揭示了额外的候选驱动,ARID1A,ROBO2,KDM6A而且PREX2[19]。这些突变与12个核心信号通路相关,包括细胞凋亡、DNA损伤、KRas信号、TGFB信号通路和表观遗传修饰,在超过三分之一的评估癌症中被发现为靶点(表2)[18,19]。值得注意的是,在染色体结构中检测到一长串不常见的变异,其中许多包含致癌基因(ERBB2, MET, FGFR1, CDK6, PIK3R3和PI3CA),这可能是可药物靶向的靶点,也意味着显著的肿瘤间和肿瘤内异质性[19]。重要的是,Waddell等.[19]表明基因组不稳定与DNA维持基因失活共分离,BRCA1, BRCA2而且PALB2,以及DNA损伤修复缺陷的突变特征。
表1。提示PDAC遗传危险因素[11]。
风险因素 |
基因 |
风险增加 |
乳腺癌和卵巢癌综合征 |
Brca2 brca1 palb2 |
2 - 5 |
家族性非典型性多痣黑色素瘤 |
CDKN2A |
47 |
Peutz-Jeghers |
STK11 / LKB1 |
132 |
遗传性非息肉性结直肠癌 |
MMRs (MSH2, MLH1, PMS1, PMS2, MSH6) EPCAM |
8.6 |
家族性腺瘤性息肉病 |
APC |
4.5 6 |
遗传性胰腺炎 |
PRSS1, SPINK1 |
69 |
囊性纤维化 |
雌性生殖道 |
3.5 |
Li-Fraumeni |
TP53 |
7.3 (%) |
Ataxia-telangiectasia |
自动取款机 |
增加 |
非o型血型 |
|
1.3 |
家族性胰腺癌 |
未知的 |
9(1罗斯福) 32(3罗斯福) |
(富兰克林·罗斯福,一级亲属)
表2:大多数胰腺癌的核心信号通路和过程发生了基因改变[18,19]。
信号通路和过程 |
基因改变的基因 |
喀斯特信号 |
Kras map2k4, rasgrp3, prex2 |
Wnt / Notch信号 |
Myc, ppp2r3a, wnt9a, gata6, tcf4, map2, tsc2 |
依赖于gtpase的小信号(KRas除外) |
Aghgef7, aghgef9, cdc42bpa, depdc2, plcb3, plcb4, rp1, plxnb1, PRKCG |
转化生长因子-β信号 |
Tgfbr2, bmpr2, smad4, smad3 |
c-Jun n端激酶信号 |
Map4k3, tnf, atf2, nfatc3 |
整合素信号 |
Itga4, itga9, itga11, lama1, lama4, lama5, fn1, ilk |
刺猬信号 |
Tbx5, sox3, lpr2, gli1, gli3, boc, bmpr2, crebbp |
入侵管制 |
Adam11, adam12, adam19, adam5220, adamts15, dpp6, mep1a, pcsk6, apg4a, prss23, robo2 |
同质性细胞粘附 |
Cdh1, cdh10, cdh2, cdh7, fat, pcdh15, pcdh17, pcdh18, pcdh9, pcdhb16, pcdhb2, pcdhga1, pcdhga11, pcdhgc4 |
G1/S相变的调节 |
Cdkn2a, fbxw7, chd1, apc2 |
DNA损伤控制 |
Ercc4, ercc6, ep300, ranbp2, tp53 |
细胞凋亡 |
Casp10, vcp, cad, hip1 |
KRas信号通路
致癌基因突变喀斯特在99%的Pan1N-1病变中发现最早可检测到的基因改变,在近100%的PDAC中发现[21,22]。突变喀斯特将KRas锁定在永久激活状态,导致下游信号通路的构成激活,包括B-raf/MAPK/ERK(细胞外信号调节激酶),磷酸肌醇-3激酶(PI3K)/3-磷酸肌醇依赖蛋白激酶1 (PDK1)/AKT激酶和Ral鸟嘌呤核苷酸交换因子[22]。喀斯特驱动的小鼠研究表明致癌喀斯特(喀斯特G12D或喀斯特G12V)对于PDAC和转移性病变的起始、进展和维持都至关重要[23,24]。在肿瘤抑制基因中引入失活突变CDKN2A,TRP53,DPC4 / SMAD4或者激活突变BRAF,大大加快了PanINs和PDAC的开发喀斯特驱动的基因工程动物[25,27]。然而,致癌的存在喀斯特表明KRas单独激活不太可能单独促进[28]的癌变。炎症刺激和KRas信号的相互作用足以驱动全谱PanIN、粘连增生和侵袭性PDAC[29]的发展。从力学上看,喀斯特信号上调Hedgehog信号、炎症介质(如核因子- κ - b (NF -κB)、环氧合酶2 (COX-2)、转录激活因子3 (STAT3))的生成,这些介质可介导上皮细胞及其周围微环境之间的旁分泌相互作用[29,30]。
值得注意的是,在缺氧/营养匮乏的条件下,致癌喀斯特通过重编程肿瘤代谢使突变细胞获得选择性优势,以维持生长和存活[31]。通过这样做,KRas信号通路促进糖酵解、谷氨酰胺驱动的氧化磷酸化、自噬和大胞饮作用。kras驱动的自噬在细胞内提供额外的营养来源,并保护细胞免受ros介导的损伤,提供抗应激能力[32]。
IGF /胰岛素轴
回顾能量密集饮食、饮食相关代谢障碍(如肥胖和2型糖尿病)和PDAC发病风险增加之间的联系,极有可能外周胰岛素抵抗、胰岛素代偿性生产过剩和胰岛素样生长因子1 (IGF-1)生物利用度增加是PDAC的重要因素。上调的IGF-1信号通路通过诱导葡萄糖摄取、分化、迁移、细胞增殖和存活[33]参与PDAC的发展和进展。在原位模型中,自分泌IGF-1/IGF-1R信号通路导致发生在致癌KRas/B-raf/ERK下游的PI3K/AKT信号的激活,在胰腺肿瘤启动[34]中发挥作用。反过来,AKT信号通过上调IGF-IR表达[35]促进PDAC细胞的侵袭性。v-AKT胸腺瘤病毒致癌基因同源物2 (AKT2)在10% - 20%的pdac[36]中观察到基因扩增、过表达和激活。AKT的激活由磷酸酶和张力同源物(PTEN)负向控制,PTEN可能由于启动子[37]的高甲基化而下调。分别,胰腺特异性缺失1个拷贝PTEN会迅速加速吗喀斯特G12D驱动的PDAC[38]。在大多数PDAC病例的进展过程中,关键肿瘤抑制基因p53的失活也导致IGF-1/AKT/mTOR通路[39]的上调。除了促进生长的信号,mTOR还介导负反馈循环,通过抑制胰岛素受体底物(IRS-1)的激活和表达来抑制信号。IRS-1将胰岛素和IGF-1的信号传递到PI3K/AKT和ERK/MAPK通路[40]。
IGF-1R可能与胰岛素受体(IR)、G蛋白偶联受体(GPCRs)、上皮生长因子受体(EGFR)、MET相互作用,从而促进PDAC[41-44]。值得注意的是,胰岛素/IGF-1受体(IR/IGF-1R)系统在PDAC的发育[41]中起着关键作用。由于其高度同源性,IR与IGF-1R[41]形成杂交种。胰岛素受体异构体A和IGF-1R杂交体结合胰岛素和igf具有类似的亲和力,特别是在高浓度胰内胰岛素时[41,45]。通常在胎儿组织中发现的胰岛素受体亚型A (IR-A),从增生性病变阶段逐渐增加到PDAC[45]。IR-A的过表达通过多种机制加速生长途径,包括;i) IGF-II与IR-A结合,ii) IGF-II与IR-A/IR-B杂交体结合,iii) IGF-II与IR-A/IGF-IR[45]结合。
STK11 / LKB1-AMPK通路
丝氨酸/苏氨酸激酶11,肝激酶B1 (STK11/LKB1)失活突变出现在家族性和散发性PDAC中[12,47]。在小鼠模型中,LKB1突变被证明与喀斯特通过抑制p21依赖的生长抑制机制[47]促进PDAC。在代谢应激条件下,LKB1通过amp活化蛋白激酶(AMPK)的磷酸化起作用,AMPK是中枢代谢传感器[48]。当ATP浓度降低,5'AMP浓度升高时,细胞内钙含量增加,AMPK也会被激活2 +和/或药物(如二甲双胍)给药[49]。众所周知,激活的AMPK可以抑制mTOR信号[49]。
EGF/EGFR信号通路
EGFR家族成员诱导细胞增殖、血管生成、运动、侵袭、转移、生存和上皮-间质转化(EMT),并通过激活下游信号通路,包括KRas/B-raf-MEK/ERK、JAK/STAT、PI3K/AKT/mTOR和Ca,减少细胞凋亡2 +CaM信号[50,51]胰腺癌中EGFR及其配体共表达增加与更多的肝转移和较差的预后[52]相关。
TGFB/SMAD信号通路
转化生长因子β (TGFB)信号通路的个别成分在PDAC中被解除调控,包括TGFBR2和SMAD4/在胰腺癌中缺失4号位点(DPC4)基因和TGFB的过表达[53,54]。TGFB信号通路与癌细胞增殖、分化、侵袭、肿瘤血管生成、细胞外基质沉积和抗肿瘤免疫[53]的抑制有关。失活的SMAD4肿瘤抑制基因存在于约60%的pdac中,尤其是高级别PanIN-3[53,54]。的损失SMAD4导致PDAC发生广泛转移,[54]生存率降低。
细胞周期控制和DNA损伤反应途径
控制细胞周期和DNA损伤反应的基因突变与PDAC有关。这些突变可以遗传自父母,也可以通过致癌物获得,如香烟烟雾致癌物,或偶然。缺乏DNA损伤反应和细胞周期检查点导致突变积累,基因组不稳定和不受控制的增殖。
周期蛋白依赖性激酶抑制剂2A(CDKN2A)在约95%的pdac中失活,绝大多数的改变早在PanIN-2阶段就发生了[18,54]。CDKN2A有几个可选的剪接位点可产生转录物变体,包括周期蛋白依赖性激酶抑制剂“p16 (p16INK4a)”和p53激活剂“交替开放框架”(ARF, p14ARF)[55]。p16抑制视网膜母细胞瘤(RB)的磷酸化,从而阻止进入细胞周期的S (DNA合成)阶段[55]。因此,p16的缺失导致G1/S过渡失控和细胞分裂失控。
失活突变TP53已在高级别(PanIN-3)原发性PDACs和50%的>病例的转移性病变中检测到[18,55]。TP53[55]参与细胞周期阻滞、凋亡、衰老、DNA修复和代谢,以维持基因组的稳定性。在应激,特别是基因毒性应激下,突变性共济失调毛细血管扩张症(Ataxia毛细血管扩张症,ATM)和p14都能激活和稳定p53东盟地区论坛[55]。除了p53, ATM还激活了其他几个关键蛋白,如BRCA1、范可尼贫血组D2蛋白(FANCD2)和丝氨酸/苏氨酸蛋白激酶CHK2,以启动DNA损伤检查点的激活,导致细胞周期阻滞、DNA修复或凋亡[55]。ATM和p14都丢失了东盟地区论坛功能在PDAC[11,55]中有很好的记录。突变体TP53由于p21无法与DNA结合,因此无法刺激p21肿瘤抑制蛋白的产生,在30-60%的PDAC病例中检测到p21表达的缺失[47,56]。
在家族性PDAC病例中已经报道了BRCA1和BRCA2的种系突变[12,57]。BRCA1/2的正常功能需要与一个修复蛋白RAD51和一个被称为“BRCA1/2的伴侣和定位者”(PALB2)的伴侣形成复合体[58,59]。这个复合体的协同同源重组(HR)由一系列相互关联的通路组成,在DNA双链断裂(dsb)的修复中起作用。在…面前BRCA如果由Poly(adp -核糖)聚合酶(PARP)调控的碱基切除修复(BER)挽救途径不受影响,它就能维持基因组的稳定性[60]。这些途径的缺陷导致DNA损伤积累、基因组不稳定、抗辐射的DNA合成、胞质分裂受损、增殖阻滞、对DNA损伤剂过敏和细胞死亡[57-60]。
种系突变错配修复(MMR)已在家族性PDAC[12]病例中报道。MMR基因在生物学上具有高度保守性,在维持基因组稳定性方面起着关键作用[12,61]。MMR功能缺陷与全基因组不稳定、对化疗药物的耐药性和减数分裂异常相关,所有这些都可能导致侵袭性肿瘤表型,包括早发性PDAC[11,61]。
酸性微环境是肿瘤组织促进侵袭表型的主要特征。据描述,在PDAC中,致癌性KRas信号和缺氧都增加了“糖酵解开关”,导致乳酸的产生和输出增加,这归因于酸性微环境的形成[31,32]。除乳酸外,CO过量2由于碳酸酐酶9 (ca9)在增生的导管上皮和PDAC中过度表达,它催化二氧化碳的可逆水化,生成碳酸氢盐和质子(CO2+ H2O↔hco3.-+ H+)[62]。这个反应发生在酶的细胞外区域,碳酸氢盐通过特定的转运蛋白进入细胞质以缓冲细胞内的pH值,而H+残留在细胞外空间,降低细胞外pH[62]。因此,ca9有助于产生和维持有利于肿瘤生长和存活的细胞内碱性pH值[63]。同时,ca9参与细胞外酸性空间的生成,促进细胞入侵[64]PDAC细胞中酸挤压增加的其他介质包括Na+/小时+交换剂(如NHE1),各种HCO3−转运蛋白(如碳酸氢钠共转运蛋白4/SLC4/NBC)、H+泵(如v型H+- atp酶)和乳酸- h+共转运蛋白(如单羧酸转运蛋白(MCTs))在PDAC中上调(图4)[65]。特别是,EGF/KRas/NHE1途径通过局部细胞外酸化和诱导具有较高转移潜力的好氧糖酵解表型参与了PDAC的早期进展[66]。其他具有质子导电性的离子通道家族也与PDAC的发病机制有关。其中,在增殖迁移、侵袭转移中起作用的“美拉斯丁相关”型(TRPM)、8型(TRPM8)、7型(TRPM7)的TRP阳离子通道和瞬时受体电位规范亚型1(TRPC1)通道[67,68]。
囊性纤维化跨膜电导调节器(CFTR)功能障碍也会导致腺泡腔内酸化。CFTR作为阴离子转运体,促进导管HCO3.-分泌[69]。突变雌性生殖道导致Cl出现故障-再循环和HCO3 -分泌,降低腺泡腔内pH值,抑制分泌颗粒蛋白的腺泡内吞作用,降低分泌管腔内蛋白的溶解度。这会通过粘液和消化酶阻塞导管,随后导致腺泡破坏、炎症和纤维化[69]。因此,这些因素中的一个或多个可能促进急性和慢性胰腺炎的发展,PDAC[12]。因此,杂合突变也就不足为奇了囊性纤维化跨膜传导调节器(雌性生殖道)基因与胰功能不全、CP、家族性PDAC和早发性PDAC相关[12,69]。
表观遗传改变有助于PDAC的发展。影响基因表达的主要表观遗传机制包括DNA甲基化、组蛋白修饰和微rna表达。基因表达模式的改变可导致致癌途径的激活、肿瘤抑制因子的沉默和致癌基因的激活,从而导致肿瘤的改变。毫不奇怪,从PanIN病变到侵袭性PDAC的表观遗传调控几乎影响了所有细胞功能,如细胞周期控制(如p16)、dna损伤反应(如p16)、一种(人类mutL同源物1)),增殖(例如,RUNX3(Runt相关转录因子3),避免细胞凋亡(例如,RPRM持续血管生成(如miR-34a),迁移和侵袭(如S100钙结合蛋白A4) (S100A4))(70 - 72)。表3总结了目前有关PDAC表观遗传改变的文献。
表3.PDAC发病机制中一些常见的表观遗传改变综述[70-72]。
表观遗传变化 |
基因的影响 |
已知或预测函数 |
DNA甲基化 |
CDKN2A |
细胞循环控制 |
|
CCND2 |
细胞循环控制 |
|
一种 |
dna损伤反应 |
|
RPRM |
p53诱导的细胞周期阻滞 |
|
BNIP3 |
缺氧引起的细胞死亡 |
|
RASSF1 |
细胞生长抑制剂 |
|
RUNX3 |
增殖和凋亡的调控 |
|
ZEB2 |
生长和发展的调节器 |
|
UCHL1 |
增殖和分化的调节 |
|
SPARC |
细胞周期进展抑制,细胞基质相互作用 |
|
MIR148A |
增殖,菌落形成 |
|
背景 |
信息联系 |
|
CLDN5 |
信息联系 |
|
SFRP1 |
Wnt信号的制造者 |
|
NPTX2 |
神经传输 |
|
PENK |
神经肽的前兆 |
|
ppENK |
Neutopeptide发射机 |
|
|
|
DNA甲基化 |
S100P |
细胞周期的进展和分化 |
|
LCN2 |
上皮分化 |
|
MIR200 |
EMT |
|
MSLN |
细胞表面抗原/细胞粘附 |
|
CLDN4 |
细胞粘附和入侵 |
|
PSCA |
细胞表面抗原/细胞分化 |
|
S100A4 |
运动性,侵袭性,微管蛋白聚合 |
|
SERPINB5 |
调节细胞运动和细胞死亡 |
|
TFF2 |
分泌多肽/上皮修复 |
microrna |
表达水平 |
目标基因 |
对细胞功能的影响 |
致癌大鹏展翅 |
↑miR-21 |
PTEN |
↑增殖,入侵,药物抗性 |
|
↑mir - 221 |
CDKN1B |
细胞周期进展,化学敏感性 |
|
↑miR-10a |
Hoxa1 hoxb1 3 |
↑侵袭转移 |
|
↑mir - 224 |
CD40 |
↑入侵,转移 |
|
↑mir - 155 |
TP53INP1 |
↓细胞凋亡 |
|
|
|
|
Tumour-suppressive大鹏展翅 |
↓Let-7 |
喀斯特 |
↑扩散 |
|
↓mir - 421 |
Smad4 |
增殖,菌落形成 |
|
↓miR-34a |
TP53 |
↓细胞凋亡与DNA修复,↑细胞周期进展与血管生成 |
|
↓miR-34 |
bcl - 2,切口 |
↑增殖,↓凋亡,↑侵袭, |
|
↓mir - 143 |
Get1, get2, kras |
增殖,入侵,迁移 |
|
↓mir - 146 a |
表皮生长因子受体 |
↑入侵 |
|
↓mir - 200家庭 |
Zeb1, sip, ep300 |
↑EMT,转移 |
(↑,增加;↓下降)
癌症干细胞(CSCs)被描述为表型独特的癌细胞,具有增强的肿瘤启动潜力、自我更新和再现原始肿瘤细胞异质性的能力[73]。胰腺癌干细胞(PanCSCs)占所有胰腺癌细胞的0.5% ~ 1.0%,表达表面标记CD44+, CD24+、上皮特异性抗原(ESA)+[73]。CD44+CD24+,欧洲航天局+PancSCs强烈上调Sonic hedgehog (Shh)配体和多梳组(PCG)基因家族成员BMI-1的转录水平,控制细胞命运、自我更新和多谱系分化[74]。整合EGFR和Hedgehog信号可诱导SOX2、SOX9、CXCR4、成纤维细胞生长因子-19 (FGF-19)的表达,这些是肿瘤启动所必需的[75]。此外,据报道,MET、Notch、Wnt/catenin -1、PI3K/AKT/mTOR和TGFB信号通路也参与了PanCSCs生物学[76,77]。
PDAC也含有1% - 3%的CD133+这种癌细胞对化疗具有高度耐药性,并与CD44部分重叠+CD24+,欧洲航天局+PancSCs[78]。一些CD133+癌细胞也表现出高表达CXC趋化因子受体(CXCR4),一种基质衍生因子1受体(sdf1 /CXCL12)[78]。重要的是,来自晚期转移性疾病患者的人胰腺癌组织标本的侵袭面表达了高水平的CXCR4+,表明CD133的作用+和趋化因子受体CXCR4+转移中的细胞[78]。因此,阻断CD133+/CXCR4+细胞可防止小鼠移植瘤转移[78]。SDF 1在肺、肝、骨髓和淋巴结中强表达,这些部位通常受胰腺癌转移影响[79]。缺氧微环境还通过诱导EMT信号和ca9的表达增强了PanCSCs获得迁移能力[80]。事实上,缺氧通过上调自噬相关基因的表达增强了panscs的克隆性生存和迁移[81]。与kras表达突变的胰腺癌细胞相比,PanCSCs细胞更少地依赖糖酵解,更多地依赖线粒体呼吸来产生能量,因此,它们产生更多的活性氧(ROS)[82]。因此,上调自噬提供了保护和抵抗内源性和外源性应激,如ROS,营养剥夺和缺氧。
粘连增生构成了PDAC的一个动态腔室,在肿瘤的形成、进展和转移中起着至关重要的作用,甚至可能在正常上皮生理存在的情况下负责肿瘤发生的启动。癌细胞与基质细胞相互作用,调节细胞外基质(ECM)的产生和组成,增加炎症细胞的招募,促进胰腺癌星状细胞的增殖和活化(图4)。
在PDAC中,免疫反应主要由免疫抑制和促肿瘤因子组成,这些因子甚至在早期就存在[83,84]。体内谱系追踪实验表明,炎症细胞和具有干细胞特性的细胞之间的旁分泌相互作用诱导EMT和转移到肝脏,这一过程甚至发生在肿瘤被标准组织学检测到之前[85]。尽管临床和动物模型都提供了炎性间质引发PDAC发展并允许其进展的有力证据,[83,84]但仍有许多证据支持正常胰腺间质抑制胰腺肿瘤形成的观点[86]。例如,来自脂肪组织的人基质细胞在体外和体内都能强烈抑制PDAC增殖,并诱导肿瘤细胞死亡[87]。
研究表明,肥胖、遗传因素(如基因突变等)引起的长期进行性炎症PRSS1或SPINK1)、生活方式因素(例如饮酒和吸烟)或其他肿瘤相关因素(例如基因突变)喀斯特),能够增强胰腺肿瘤[32,87,88]。
一项调查肥胖对胰腺癌发生的影响的研究表明,高脂肪饮食通过COX-2激活KRas信号,导致胰腺炎症和纤维化,并随后产生PanINs和PDAC[30]。事实上,与烟草有关的致癌物,包括亚硝胺、多环芳香烃及其代谢物,会导致喀斯特而且TP53促进胰腺炎症和PDAC[20]。致癌KRas通过PanIN细胞上调促炎细胞因子(如白介素(IL)-6、IL-11、肿瘤坏死因子(TNF)- α、IL-1α),向促肿瘤发生微环境发出信号[88]。这些细胞因子通过自分泌方式激活JAK2/STAT3和NF-κB通路诱导PanIN细胞增殖和存活,并招募免疫细胞(尤其是髓系细胞)[88]。最近的研究发现PanINs中存在b细胞亚群,促进促肿瘤(TH2型)巨噬细胞表型(肿瘤相关巨噬细胞- tam -)导致免疫抑制和PDAC进展[89]。招募免疫细胞分泌的免疫调节介质和生长因子(如IL-35、IL-6、IL-11、tnf - α、il -1 α、IL-10、il -1 β、IL-2、ROS、EGF、TGFB、HH和MMPs),以创建一个正反馈循环并抑制细胞毒性T细胞应答(CTL)[88-90]。这些细胞因子促进了PanIN和PDAC细胞的EMT、增殖和存活,并抑制癌基因诱导的衰老[87,88]。重要的是,TNF-alpha刺激上皮细胞中ROS的积累,引起DNA损伤和基因组不稳定,从而促进致癌突变[30,91]。此外,细胞因子与KRas协同激活Notch和Hedgehog信号以加速PDAC的发育[30,95]。髓系衍生抑制细胞(MDSCs)是一种免疫抑制细胞类型,可抑制CTL反应并诱导调节性T细胞(treg)的发育[84,91,92]。PDAC中的t淋巴细胞以treg为主,参与免疫反应的抑制,在PDAC患者的血液和胰腺组织中显著增加[92,93]。treg和MDSCs的积累与疾病进展呈正相关,与患者生存期负相关[92]。值得注意的是,肿瘤来源的乳酸生成增加了抑制自然杀手(NK)细胞毒性的MDSCs的数量[94]。PDAC细胞还表达粒细胞巨噬细胞集落刺激因子(GM-CSF)、IL-10、-4、-6、TGFB等多种因子,这些因子反过来抑制树突状细胞(DC)的成熟,从而限制t淋巴细胞的增殖[93,95]。 Accordingly, decreased circulating DCs and decreased NK activity are observed in PDAC patients [96]. Indeed, PDAC cells can induce apoptosis of infiltrating T cells by secretion of Fas ligand as well as by downregulating expression of human leukocyte antigen (HLA) I molecules and Fas signalling, thus blocking and evading the immune response at the tumour site [97,98]. In fact, PDAC cells express a variety of cancer-associated antigens that can potentially be recognised by T lymphocytes [97,99,100]. Several studies have revealed that tumour-specific Cytotoxic T Lymphocytes (CTL/CD8+ T) precursors present in peripheral blood and bone marrow of pancreatic cancer patients [99,100] Indeed, the infiltration of the tumour by effector CD8+, CD4+ T cells and dendritic cells was found to be a good indicator of the patient’s outcome after surgical treatment [100].
胰腺星状细胞(PSCs)(也称为肌成纤维细胞或癌症相关成纤维细胞)是PDAC基质中的主要间充质型细胞[101,102]。在正常、健康的胰腺中,它们少量处于静止状态,位于外分泌胰腺的腺泡周和导管周区域[101]。它们的细胞质中含有典型的含维甲酸的脂肪液滴,有丝分裂指数低,ECM合成能力低[101]PSCs被一系列因素激活,包括促炎细胞因子、生长因子、氧化应激、毒素(例如,酒精及其代谢物、内毒素)、缺氧、间质压升高、高脂肪饮食和高血糖[30,102]。激活后,它们从静止状态转变为激活的肌成纤维细胞状态[101,102]。激活的PSCs失去脂肪滴(含视黄酸),表达α-平滑肌肌动蛋白(α-SMA)合成生长因子(如TGFB, VEGF, HGF, tnf - α, PDGFB)和炎症细胞因子(如IL-6, il -1 β)以及过量的ECM蛋白(包括胶原蛋白,层粘连蛋白,纤维连接蛋白和骨膜素),形成纤维组织[16,103-105]。一旦被激活,PSCs可以通过形成自主反馈回路来保持自身活性,并促进PDAC细胞的增殖、迁移、侵袭、转移、EMT和存活[16,103-105]。反过来,肿瘤细胞产生生长因子诱导PSCs细胞分泌ECM蛋白[105]。PSCs还调节基质的再吸收和周转,主要通过产生mmp[102]。PSCs的主要产物骨膜素和胶原蛋白在PanIN、IPMNs和PDAC的基质中表达增加,其表达与恶性转化的阶段平行增加[15,104-106]。
PSCs通过分泌细胞因子和VEGF促进MDSCs增殖和激活,损害T细胞存活,分别通过CXCL12/CXCR2和CXCL12/CXCR4招募Treg和隔离ctl,阻碍其与肿瘤细胞的接触,在介导PDAC中的免疫抑制微环境中发挥重要作用[107-109]。因此,t淋巴细胞被显示包围在胰腺病变周围,并且在纤维化间质组织中比在PDAC的上皮内区域中更常见[109]。PSCs释放IL-33激活肥大细胞产生促炎细胞因子,mmp的产生促进PDAC进展[91]。此外,PSCs细胞分泌SDF 1可通过激活SDF 1/CXCR4轴诱导癌细胞侵袭[110]。SDF 1的这些梯度可能会吸引PSCs和PDAC细胞,并调节特定转移位点的增殖和侵袭[110]。事实上,PSCs伴随癌细胞转移到远处的转移位点,在那里它们可能促进癌细胞的播撒、生存和增殖[111]。有趣的是,天山等PDAC细胞通过PDGF、FGF2、TGFB刺激肝星状细胞的激活,从而修饰肝脏间质,使其更适合存活[112]。鉴于造血干细胞和PSCs之间的相似性,以及原发性胰腺肿瘤和相关继发性肝转移瘤之间的胶原分布模式相似,我们有理由推测造血干细胞在PDAC细胞向肝脏转移过程中发挥关键作用[113]。
最近的研究也表明PDAC的神经生长和神经周围侵犯(PNI)与PSCs有关。17在人PDAC中,粘连增生的程度与神经侵犯的程度呈正相关。炎症细胞、免疫细胞或癌细胞引起的PNI损伤神经,并引起与PDAC[17]相关的经典预后性胰腺神经性疼痛,这已得到充分证实。基于体内模型的研究,神经周围侵袭似乎是通过Sonic hedgehog (Shh)信号通路触发的,该信号通路反过来激活肿瘤微环境中基因表达谱发生改变、突变的PSCs,并导致肿瘤进展[114]。神经元的生长和伸长也受到胶原蛋白、纤维连接蛋白和透明质酸的影响,这些主要由PSCs产生[115]。此外,神经胶质来源的NGF、胆碱能和交感神经输入促进癌细胞侵袭和增殖[116,117]。
致密的富含胶原的纤维性ECM是PDAC的标志之一。尽管PDAC具有很高的转移潜力,这种致密的纤维结构似乎可以作为迁移和侵袭的屏障。矛盾的是,粘连增生反应以刺激PDAC进展和转移的方式起作用。
ECM除了富含免疫调节介质和生长因子外,还含有多种细胞-基质相互作用调节剂,包括凝血蛋白、骨膜蛋白、肌腱蛋白C (TNC)、分泌酸性蛋白并富含半胱氨酸(SPARC)、玻璃蛋白、大聚糖、胶原(主要是I型、III型和IV型)、层粘连蛋白和纤维连蛋白以及蛋白多糖和糖胺聚糖[118]。整合素和CD44信号是PDAC中细胞与ECM通信的重要手段。多个整合素亚基可以以多种组合相互作用,形成对ECM蛋白具有不同亲和力的独特受体,促进粘附、生存、生长、迁移和侵袭[119]。
除了组成外,ECM的硬度还调节肿瘤的生物学[119]。I型胶原、MT1-MMP和TGFB信号之间存在一个正反馈循环,促进粘连反应的建立和维持并支持迁移[119]值得注意的是,胶原表达的增加被用来计算每个肿瘤中基质的激活指数,该指数越高被发现与PDAC患者的预后越差呈正相关[15,16]。
除了提供信号支架外,持续的纤维形成和截留液体的粘多糖(如透明质酸)作为灌注屏障,导致高间质液,并改变血管和微毛细血管的组织和结构[120]。所有这些修饰改变了血管对营养和治疗的通透性,并导致缺氧[110]。缺氧诱导基质细胞和PDAC细胞释放血管生成因子(如VEGF、FGF、血管生成素2、骨膜抑素、COX-2、神经素-1),诱导血管生成,导致肿瘤过度生长。血管生成信号通路的激活已被发现与PDAC患者的不良预后相关,也与肝和肺转移有关[121-123]。同时,PSCs的持续激活导致ECM分子过度沉积,诱导PDAC细胞产生内皮抑素,内皮抑素是内皮增殖的抑制剂,可有效抑制血管生成[124]。因此,与预期相反,这种对微环境的操纵会压制局部促血管生成的特性,从而产生低血管微环境和肝硬化/缺氧组织[124]。这些发现可能解释了PDAC抗血管生成治疗的不足,并提出了针对肿瘤间质相互作用的新的治疗方法。
诊断
PDAC通常在疾病发展后期出现临床症状,此时肿瘤已经发展到晚期或已经扩散到胰腺以外或转移到其他器官。PDAC的症状取决于肿瘤在胰腺内的位置,以及疾病的阶段。然而,大多数症状是模糊的,可以归因于许多不同的条件,导致恶性肿瘤发现较晚。另一个导致发现晚的因素是,胰腺的功能相对不受影响,直到超过50%的组织失去功能。此外,胰腺在腹部深处的位置使初级保健医生无法对其进行体检。因此,在目前可用的诊断手段下,准确和早期发现PDAC是非常困难的。
大多数pdac位于胰腺的头部。尽管这并不一定会改变疾病的生物学特性,但身体或胰腺尾部肿瘤患者比胰腺头部肿瘤患者在解剖学上具有优势,因为他们不易发生胆道阻塞,因此不太可能需要可能增加感染风险的干预措施,特别是在接受化疗治疗时。
临床病史
常见的临床特征包括腹部持续疼痛,特别是辐射至背部的上腹痛,原因不明的体重减轻,黄疸,黏土色大便,深色尿,恶心和/或呕吐,脂肪变性,不适和凝血功能障碍[125]。起源于胰腺任何部位的PDAC可能与糖尿病的新发或恶化有关[131]。约70%的患者存在糖尿病,通常糖尿病史不超过2年[126]。随后的症状与肝转移和/或邻近器官(胃、结肠)或腹膜腔的侵犯有关,这可能导致腹水[126]。黄疸和肝功能异常也可能表明癌症已转移到肝脏[126]。偶尔,患者会出现急性胰腺炎、移行性血栓性静脉炎或高钙血症[3]。抑郁在胰腺癌患者中也很常见[127]。
实验室检查
PDAC患者的实验室检查结果通常是非特异性的。然而,初始血检通常包括全血细胞计数(CBC)、全代谢谱(CMP)、血清淀粉酶和/或脂肪酶和肿瘤标记物(癌症抗原19-9 (CA 19-9)、Du-Pan 2、癌胚抗原(CEA)、Span-1)[126]。总胆红素和直接胆红素测量以及包括血清转氨酶(AST/ALT)和碱性磷酸酶在内的肝功能检测可揭示胆道梗阻和肝转移的证据[126]。胰管阻塞或胰腺组织损伤可导致血清淀粉酶和脂肪酶水平升高[128]。CA19-9是美国食品和药物管理局批准用于胰腺癌的唯一肿瘤标志物[129]。然而,CA19-9不是PDAC的特异性肿瘤标志物,因此不应单独用于PDAC筛查,因为在其他情况下,如胰腺炎、胆结石、胆汁淤积、肝病和各种炎症性疾病,其水平可能升高[119]。此外,该检测在没有功能性Le酶的个体中无效,Le酶在CA 19-9的翻译后修饰中发挥作用[129]。
放射学
目前,没有一种单独的方法能够提供足够的敏感性和特异性,因此,在对疑似PDAC患者的术前诊断和分期中,采用了不同的成像方式和血液检测的组合。超声(US)、计算机断层扫描(CT)、磁共振成像(MRI)、正电子发射断层扫描(PET)、内窥镜超声(EUS)、内窥镜逆行胆管胰腺造影(ERCP)、磁共振胆管胰腺造影(MRCP)和多探测器排计算机断层扫描(MDCT)是目前可用的胰腺成像技术,用于胰腺局灶性病变的表征、初始分期、手术和治疗计划。以及治疗反应评估[126,130]。MDCT最好与EUS相结合,对于病灶的早期发现更为敏感,并允许相对容易地进入胰腺,使用细针穿刺(FNA)进行组织诊断,并为肿瘤分期提供进一步的重要信息[130]。
暂存
身体检查、影像学检查、实验室检查、病理报告和手术报告都用于准确地对疾病进行分期。一旦肿块被确定,FNA证实诊断,EUS可以确定肿瘤的大小,淋巴结转移的范围,并评估门静脉系统的受累程度,完成分期。目前,分期系统用于预测患者的预后或根据疾病的分期提出最佳治疗方案。对于PDAC患者的分期,不同的社会或学术实践已经开发了几种分期系统或共识声明。这些分期系统包括由美国癌症联合委员会(AJCC)、131国家综合癌症网络、132美国肝胰胆道协会、肿瘤外科学会、消化道外科学会和德克萨斯大学MD Anderson癌症中心制定的分期系统。133所有这些分期系统主要取决于肿瘤的大小、在胰腺内的位置、是否与邻近血管接触、是否延伸到胰腺外、是否存在转移性病变[131-133]。主要扩散区域为淋巴管和区域淋巴结、胰后组织连接、肝脏、腹膜、骨髓、肺和主要血管结构的局部浸润,特别是门脉和肠系膜金星树以及发情胰神经丛[134]。根据AJCC肿瘤-节点-转移(TNM)分级,该分级是基于螺旋CT对可切除性的评估,T1、T2和T3肿瘤是可切除的,而涉及肠系膜上动脉或腹腔轴的T4肿瘤是不可切除的(表4)。涉及肠系膜上静脉、门静脉或脾静脉的肿瘤被归为T3,因为这些静脉可以切除和重建[134]。强烈建议在外科、诊断成像、病理学、介入内镜、肿瘤医学和放射学方面拥有专业知识的多学科团队来确定哪些患者适合辅助治疗手术。
表4。与7th外分泌胰腺肿瘤的TNM分类
TNM分类 |
|
原发肿瘤(T) |
TX:原发肿瘤无法评估 |
T0:没有原发肿瘤的证据 |
Tis:原位癌 |
T1:肿瘤局限于胰腺,最大尺寸不超过2厘米 |
T2:肿瘤局限于胰腺,最大直径超过2厘米 |
T3:肿瘤扩散到胰腺以外,但未累及腹腔轴或肠系膜上动脉 |
T4:肿瘤累及腹腔轴或肠系膜上动脉(不可切除) |
|
区域淋巴结(N) |
MX:不能评估局部淋巴结 |
N0:无区域淋巴结转移 |
N1:局部淋巴结转移 |
|
远处转移(M) |
MX:无法评估远处转移 |
M0:无远处转移 |
M1:远处转移 |
|
举办集团 |
阶段0 |
这 |
N0 |
M0 |
局限于胰腺内,可切除 |
阶段1 |
T1 |
N0 |
M0 |
局限于胰腺内,可切除 |
阶段1 b |
T2 |
N0 |
M0 |
局限于胰腺内,可切除 |
阶段活动花絮 |
T3 |
N0 |
M0 |
局部侵入性,可切除 |
IIB阶段 |
T1 2或3 |
N1 |
M0 |
局部侵入性,可切除 |
第三阶段 |
T4 |
任何N |
M0 |
局部晚期,不可切除 |
四期 |
任何T |
任何N |
M1 |
远处转移 |
重设表PDAC
目前,对于局部可切除的PDAC患者,唯一可接受的潜在治疗方式是手术切缘阴性的完全手术切除。专家已达成共识,制定了定义肿瘤可切除性的标准,从而确定哪些患者将从手术中受益[132,135,136]。用于切除的最常见的手术方法被称为惠普尔手术(Whipple procedure),该手术将胰腺的肿瘤承载区域以及部分胃、十二指肠、胆囊和部分胆管切除,其余区域重新连接,以支持患者的消化能力[132]。
PDAC的外科治疗有限;因此,术后治疗(辅助治疗)被认为是可切除PDAC患者的标准护理。化疗和放疗的辅助治疗均可改善无病生存率和总生存率[137]。目前对于可手术切除的PDAC的辅助治疗还没有普遍的共识。基于六项针对PDAC的显著辅助剂前瞻性随机III期试验(GITSG [138] EORTC [139] ESPAC‐1 [140,141]CONKO‐001 [142]ESPAC‐3[143]和RTOG 97‐04[144])的结果,吉西他滨或5-氟尿嘧啶(5-FU)或5-FU +叶酸化疗6个月无放疗或伴放疗是标准治疗。PDAC完全切除和吉西他滨治疗后,未治疗患者的中位无病生存期分别为13.4个月和6.7个月[143]。单独使用吉西他滨通常被推荐为目前的标准辅助化疗,因为与5-FU治疗相比,吉西他滨观察到的不良反应更少,而两个治疗组之间的生存率无显著差异(切除后的中位生存期分别为23和23.6个月)[143]。
边缘可切除肿瘤的患者在适当的术前(新辅助治疗)化疗或放化疗后可手术切除[145,146]。在一份关于边缘可切除的PDAC的大型报告中,术前基于吉西他滨的方案与基于5-FU或紫杉醇的方案相比,具有较低的镜下切缘阳性率、更大的治疗效果、较低的术后局部复发率和更好的切除术OS[145,146]。事实上,生存结果被发现与辅助治疗试验中报道的结果相当[146]。虽然中位OS仍不超过2年,但5年生存率分别为40-50%和15-20%[146]。这种新辅助方法允许识别最可能从切除中受益的患者子集,这一组中位生存期有利[145,146]。
非重切表PDAC
在绝大多数病例中,PDAC患者被诊断为局部晚期、不能手术的肿瘤(约40%的病例)或转移性疾病(约40-45%的病例),这些组报告的中位生存期分别为8-12个月和3-6个月[136,147]。初始转移部位对转移性PDAC患者的预后很重要,而肝外转移患者的预后优于肝外转移患者,两者合并的患者预后最差[148]。没有远处转移证据的局部晚期PDAC患者被定义为手术不可切除,其治疗目标与转移性疾病一样,是延长生存期、缓解症状和控制疾病[149]。与最佳支持治疗相比,系统性化疗对晚期PDAC有好处,可改善症状和OS。1997年,伯里斯et al。[150]报道在晚期疾病患者中吉西他滨优于5-FU。在这项III期研究中,研究人员证实,吉西他滨和5-FU治疗组患者的中位OS持续时间分别为5.65和4.41个月(P = 0.0025),吉西他滨和5-FU治疗组患者1年生存率分别为18%和2%[150]。自1997年首次临床疗效和安全性证明以来,吉西他滨一直是世界范围内边缘性、局部晚期和转移性PDAC患者一线治疗的基石[150]。从那时起,许多II期试验报告了各种细胞毒性药物(capacitabine,顺铂,伊立替康,奥沙利铂,培美曲塞,exatecan)和靶向药物(西妥昔单抗,蒂法尼,索拉非尼,阿西替尼,贝伐珠单抗)与吉西他滨联合的有前景的活性。然而,这些组合的III期试验并没有在晚期PDAC患者中产生有意义的临床改善或生存效益[147]。一项III期研究表明,与单独使用吉西他滨相比,在吉西他滨中加入厄洛替尼可以改善OS,但这种益处很小(6.2 vs 5.9个月),并且伴随着毒性的大幅增加[151]。
2011年,PRODIGE 4试验表明,细胞毒性联合方案FOLFIRINOX(5-氟尿嘧啶、叶酸、伊立替康、奥沙利铂)显著改善中位OS至11.1个月,而吉西他滨治疗的中位OS为6.8个月[152]。然而,在中性粒细胞减少方面,FOLFIRINOX治疗导致> 3级不良事件显著增加(FOLFIRINOX 45.7%)vs。吉西他滨21.0%),发热性中性粒细胞减少症(5.4%)vs。1.2%),血小板减少症(9.1%)vs。3.6%)、腹泻(12.7%)vs。1.8%)和周围神经病变(9.0%)vs。0%),丙氨酸转氨酶升高发生率降低(7.3%)vs。20.8%)[152]。目前FOLFIRINOX治疗仅限于性能状况良好的患者,绝大多数PDAC患者仍接受吉西他滨治疗,建议将吉西他滨作为单药治疗,因为它可通过缓解症状和延长生存期(通常为2 - 3个月)提供临床益处[152,153]。为了提高耐受性和降低严重毒性的风险,剂量修正研究表明,在局部晚期和转移性PDAC患者中,5-FU丸剂和伊立替康的剂量衰减可以在不影响疗效的情况下提高耐受性[154,155]。另一项研究使用改良FOLFIRINOX方案治疗晚期非转移性PDAC患者,显示较少的血液毒性,并在新辅助治疗中保持了令人印象深刻的切除率[156]。
2012年,晚期PDAC的MPACT试验表明,与单药吉西他滨相比,nab-紫杉醇联合吉西他滨可提高有效率(单独使用7%,联合使用23%)、无进展生存期(PFS)(从3.7个月到5.5个月)和OS(从6.7个月到8.5个月)[157,158]。正如预期的那样,与吉西他滨组相比,联合组增加了血液学毒性和非血液学临床毒性,如神经病变、疲劳、脱发和粘膜炎[157,158]。nabu -紫杉醇加吉西他滨和FOLFIRINOX的毒性是相似的。然而,FOLFIRINOX方案的血液毒性和生长因子使用率较高,而nab-紫杉醇加吉西他滨显示出较高的神经病发生率[158]。与FOLFIRINOX治疗相比,在大多数晚期PDAC患者中,nab-紫杉醇加吉西他滨治疗具有良好的耐受性和可管理性[158]。
FOLFIRINOX或nab-紫杉醇+吉西他滨一线方案最重要的临床结果之一是,越来越多的患者经历了长期的疾病控制,这使他们能够接受具有临床效益的二线和维持治疗。尽管目前还没有公认的积极的二线治疗方案,但在无法切除的PDAC患者中,已有二线全身治疗的临床试验数据。例如,一项随机的III期研究证明,在吉西他滨一线治疗后,奥沙利铂和5-FU的二线化疗是一个很好的选择[159]。此外,研究表明,局部晚期和转移性PDAC主要对FOLFIRINOX治疗耐药,但对nab-紫杉醇和吉西他滨联合治疗仍有反应,且毒性可控制[160-162]。对于FOLFIRINOX进展后的晚期PDAC患者,吉西他滨单药治疗也是一个合理的二线选择[163]。最近,研究发现纳米脂质体伊立替康联合5-FU和叶酸可延长转移性PDAC患者的生存时间,且安全性可控,且之前接受吉西他滨基础治疗。在14个国家的一项全球随机、开放标签III期试验中(NAPOLI-1)[164]。使用纳米脂质体伊立替康+ 5-FU和叶酸的患者(n=117)的中位OS为6.1个月vs使用5-FU和叶酸4.2个月(n=149)。另一方面,接受纳米脂质体伊立替康单药治疗的患者(n=151)与接受5-FU +叶酸治疗的患者(4.9个月vs 4.2个月)的中位OS没有差异[164]。
维持治疗是晚期PDAC治疗的一个新时代,它可以延长疾病控制,最终改善OS。最近,Reniet al。[165]首次探讨了维护策略在PDAC管理中的作用。舒尼替尼是一种对Raf、VEGFR和c-Kit激酶有效的抑制剂,被证明在维持治疗方面很有前途[165]。在这项II期研究中,转移性PDAC、性能状态为>50%、化疗6个月后无进展的患者被随机分组观察(实验组a)或每天舒尼替尼(中位数为91天),直到进展或最多6个月(实验组B)[165]。主要毒性分级为血小板减少症、中性粒细胞减少症、手足综合征和腹泻。该研究完成了其主要终点;A组6个月的PFS为3.6%(95%置信区间(CI): 0-10.6%),为22.2% (95% CI: 6.2-38.2%;P<0.01)。虽然OS差异无统计学意义,但B组患者两年后存活的比例是A组的三倍[165]。
采用FOLFIRINOX或nab-紫杉醇+吉西他滨的联合治疗策略可有效控制疾病,两种治疗方案已迅速应用于临床实践。然而,它们不断增加的毒性、额外的支出以及适合接受这些治疗的患者人数越来越少,限制了它们的使用[153,158]。
姑息治疗
胆道梗阻、胃出口梗阻、癌症相关疼痛、营养不良、血栓栓塞性疾病、肿瘤相关疼痛和抑郁是局部晚期和转移性PDAC最需要姑息性干预的并发症。梗阻性黄疸患者的姑息治疗包括胆道旁路或胆道支架。预期寿命超过3个月的患者可接受开放式或腹腔镜胃空肠造口术,加或不加空肠造口管;肠内支架也是一种选择。然而,对于预期寿命较短或性能状况较差的患者,可以置入肠内支架[166,167]。建议口服胰酶置换治疗外分泌胰腺功能不全[168]。低分子肝素和华法林用于预防复发性血栓栓塞性疾病[166]。严重的顽固性疼痛发生在大多数PDAC患者中。持续疼痛与肿瘤大小及胰内和胰外神经浸润呈正相关[169]。阿片类镇痛药已用于慢性疼痛[170]。 Celiac plexus block has been a common procedure to alleviate pain in patients with unresectable PDAC with potentially fewer adverse events than traditional opioid management [171]. Common, side effects include urinary retention, back pain, diarrhoea, and hypotension. Serious complications such as transient or permanent paraplegia are extremely rare [171].
总的来说,这些步骤提供了生理益处,包括改善肝细胞代谢、蛋白质合成、脂肪吸收和消化以及细菌清除[166]。此外,消化不良、瘙痒、水肿和胃淤积的缓解可改善患者的身体状况和生活质量。
治疗耐药的概念
上文讨论过的许多化疗方案,近年来已经进行了试验,大多数都未能改善PDAC患者的OS。PDAC在多个方面都被公认为是一种极具挑战性的疾病。助长这种范式的是一组其他癌症不常见的潜在生物学属性。主要原因有:i)肿瘤内部、肿瘤间的遗传和表观遗传异质性导致多重分子畸变,ii)信号通路和组成活性分子之间的交叉对话和反馈机制,iii)结缔组织增生反应,iii)免疫系统的逃避,iv) PanCSCs-所有这些都有助于PDAC的生物学和临床侵袭性。此外,临床试验的解释,以及围绕患者选择的实践和伦理并发症是最有可能导致试验失败的原因。
肿瘤内部和肿瘤间的遗传异质性是PDAC获得越来越强的侵袭性和对治疗的耐药性的机制之一。根据一项分子谱分析研究,比较了PDAC和对照患者样本之间最显著差异表达的基因/通路,表明PDAC个体间异质性的基因/通路之间几乎没有重叠[172]。此外,一项基因谱研究表明,同一患者的肿瘤内和转移之间都存在显著的异质性[80,172]。重要的是,遗传异质性主要发生在癌症发展的早期,并在癌症扩散后持续存在,导致不同转移之间持续的、平行的和事件收敛进化[80,172]。
一项有趣的研究研究了PDAC的发展和进展,该研究基于一个计算模型,该模型综合了从正常细胞到癌变细胞最后到转移细胞的体细胞改变数量和细胞的相对增殖率,提出PDAC从启动到患者死亡有很长的潜伏期,大约为几十年[173]。这一发现表明转移的发展有很长的潜伏期,因此早期诊断和治疗有很大的机会[80]。另一方面,非常小或临床上无法检测到的原发性肿瘤患者仍有很高的转移风险。实验数据表明,肿瘤前细胞的扩散是PDAC的早期事件,甚至在标准组织学检测到癌之前[80]。这些发现提醒我们,除了遗传异质性外,持续变化的微环境信号对癌细胞施加不同的选择压力,导致表型和功能异质性。因此,癌细胞的显著多样性和适应性阻碍了PDAC的治疗。此外,在治疗过程中获得新生遗传病变和治疗诱导的耐药小亚群细胞的选择,这些细胞本质上不敏感,并已经存在于原始肿瘤中,使耐药问题更加具有挑战性。
PDAC报道了吉西他滨在患者间和肿瘤内的传递和代谢的广泛异质性[174,175]。吉西他滨是一种前药,需要核苷转运体、平衡核苷转运体(ENT1)和集中核苷转运体(CNT)才能进入细胞内。在进入细胞后,它被脱氧胞苷激酶(dCK)和胸苷激酶2 (TK2)的后续磷酸化事件激活。作为脱氧胞苷类似物,活化的吉西他滨(三磷酸-吉西他滨(dFdCTP))通过抑制核糖核酸还原酶(rpm1, RRM2)阻断DNA合成并进入DNA和RNA,从而阻止生长和启动凋亡[175]。胞苷脱氨酶(CDA)的脱胺作用使细胞内吉西他滨大部分失活[174]。基于吉西他滨转运和代谢的获得性耐药与ENT1、CNT1、CNT3、dCK和TK2缺乏以及RRM1和RRM2过表达相关[174-179]。miR-21的表达增加和miR-200的表达减少有助于吉西他滨的化疗耐药,并增加癌细胞的侵袭性[180]。此外,在胰腺癌发生过程中,p53功能的丧失、抗凋亡蛋白(Bcl家族蛋白)、NF-κB和缺氧诱导因子1- α的表达增加以及SRC激酶、EGFR、STAT3、PI3K/AKT、Notch和MAPK通路的更高激活导致吉西他滨耐药[178-184]。同样,在延长5-FU治疗过程中,激活包括抗凋亡、SRC激酶和EGFR/AKT在内的分子级联反应可使PDAC细胞存活[180,186]。5-FU是口服卡培他滨的活性形式,可以抑制胸苷酸合成酶(TS)并将其代谢产物结合到RNA和DNA中。 Not surprisingly, deficiency in 5-FU transporters (e.g. human concentrative nucleoside transporter (CNT1)) and upregulation of multiple drug resistance-transporters (MRPs) over the course of 5-FU treatment reduces the accumulation and cytotoxicity of 5-FU [179,187,188]. Thus, the molecular heterogeneity present within the tumours provide a fitness advantage under selective forces.
细胞毒性药物的非特异性、狭窄的治疗指数和显著的毒性导致了靶向治疗的发展,其目的是干扰确定的分子过程。尽管已经取得了令人印象深刻的结果,并且在许多患者中观察到肿瘤回归,但组成性活性分子、正反馈环和受体串扰限制了靶向药物的疗效。例如,作为PDAC启动的主要驱动因素和多种信号通路的中心中介,致癌KRas仍然是药物开发人员最重要的治疗靶点。然而,所有直接干预KRas酶活性的临床尝试都失败了,因为突变将KRas锁定在永久活性状态[189]。因此,人们试图抑制KRas,而不是抑制酶活性,主要是通过阻断FTase活性抑制必要的翻译后修饰[190]。然而,一项II期研究显示,FTase抑制剂15777在晚期PDAC患者中没有表现出单药活性[190]。此外,在一项针对晚期PDAC患者的III期试验中,与吉西他滨单药治疗相比,蒂法尼布(一种FTase抑制剂)和吉西他滨联合治疗的疗效-毒副关系不佳,生存期没有改善[191]。有人认为,保留KRas功能的代偿性香叶酰转移酶活性的增加可能是FTase抑制剂临床失败的原因之一[192]。结论必须是,在目前,KRas应被认为是一个不可服用的靶标[22]。
KRas的组成活性形式也阻碍了egfr靶向治疗,这在非小细胞肺癌和结肠癌模型中已得到证实[193,194]。此外,EGFR/MET、EGFR/IGFR之间的交叉作用,构成了激活EGFR基因突变以及EGFR和受体酪氨酸蛋白erbB-2 (ErBB2)的受体异二聚,在获得对EGFR抑制剂(如厄洛替尼)的耐药性方面发挥了重要作用[179,195]。此外,PTEN表达缺失与AKT激活增加相关,AKT是EGFR的下游效应因子,最终对厄洛替尼产生耐药性[196]。
关于IGF-1R信号在PDAC中的核心重要性,似乎IGFR级联可能是一个潜在的很好的治疗靶点。然而,尽管IGFR抑制剂在体外已被证明可以抑制PDAC癌细胞的生长,但在临床试验中使用IGF-1R阻断抗体(如ganitumab)的效果很大程度上令人失望[197]。IGF-1R靶向治疗失败的最有效的候选者是与IR[46]的串扰。因此,IGF对ir的激活绕过了IGF- 1r及其抑制[46]。其他一些因素可能导致对igf - 1r靶向治疗的耐药性,包括下游效应分子的本构激活(例如,突变体)BRAF和变异喀斯特)以及受体与其他膜受体(包括MET和EGFR)的串扰[43,44,46]。
当二甲双胍被用来抑制mTOR信号时,也得到了令人失望的结果。二甲双胍是线粒体呼吸链复合体I的特异性抑制剂,通过减少ATP的产生促进lkb1依赖性的AMPK激活[198]。然而,LKB1基因的突变和多态性降低了AMPK的激活,并对二甲双胍不耐受,导致mTOR过度激活[198]。有趣的是,虽然mTOR的选择性抑制剂雷帕霉素抑制了IRS 1,但它会增加PDAC细胞中PI3K/AKT的激活[198]。另一方面,雷帕霉素对PDAC细胞中ERK的激活没有任何刺激作用[197]。相反,mTOR的活性位点抑制剂(KU63794和PP242)会抑制AKT的激活,而PDAC中的ERK激活会增加[198]。这些结果暗示mTOR抑制剂的治疗效果会因其他上游途径的激活而减弱,因此,在使用mTOR抑制剂进行PDAC治疗时,抑制反馈回路应该是一个主要考虑因素,以平衡mTOR的抗增殖作用。
异质细胞群之间的动态通信、基质和环境选择压力驱动治疗耐药。异常情况下,PDAC基质含量丰富,血供不足,作为药物屏障,导致全身治疗失败。当间质压降低,从而影响肿瘤血管的收缩和由此产生的血液供应时,吉西他碱的活性有可能通过使用聚乙二醇化的人重组PH20透明质酸酶诱导肿瘤血管的再扩张而增强,如异种移植的PDAC模型[199]所示。值得注意的是,吉西他滨和PEGPH20联合治疗可提高中位生存期并降低转移负担[199]。此外,使用IPI-926靶向Shh通路增加了肿瘤内血管密度和吉西他滨的肿瘤内浓度,导致癌细胞破坏增加[200]。因此,使用小分子拮抗剂或Shh配体阻断抗体5E1的本构刺猬(Hh)通路阻断也被报道可以抑制人胰腺异种移植模型中的远处转移[199,200]。然而,HH途径拮抗剂联合细胞毒性化疗的临床试验结果令人失望[201-203]。李et al。[204]在三种不同的基因工程小鼠模型中发现,对HH途径活性的遗传和药物抑制实际上加速了PDAC的进展。此外,他们还报道了通路活性的急性调节调节上皮和间质成分之间的平衡,抑制导致粘连增生的抑制和上皮成分的加速生长,激活导致间质增生和肿瘤上皮细胞的生长减少[204]。考虑到基质在肿瘤病理中的强大影响,ECM/整合素信号通路保护肿瘤细胞免受药物诱导的凋亡就不足为奇了[205,206]。因此,一项荟萃分析显示,与整合素介导的细胞粘附和耐药途径相关的基因在PDAC中经常过表达[206]。除了ecm调节的化疗耐药外,PSCs还可以直接影响癌细胞对化疗的反应。例如,PSC分泌物已被证明通过增强炎症信号[206]和CXCL12/CXCR4轴[208]促进化疗耐药。
最后,值得注意的是,免疫治疗方法,如免疫检查点抑制剂,如以靶向t淋巴细胞调节剂细胞毒性t淋巴细胞蛋白4 (CTLA 4)和程序性细胞死亡1 (PD 1)的抗体为特征的免疫检查点抑制剂,已在多种实体肿瘤中显示出疗效,并已获得美国食品和药物管理局(fda)的批准[209]。然而,由于PDAC通常被认为是一种非免疫原性恶性肿瘤,专注于克服t细胞免疫检查点的免疫治疗方法尚未被发现在胰腺癌的管理中是成功的[206,209]。
最近,人们的注意力集中在针对PanCSCs以寻找更好的方法来对抗PDAC,因为与其他癌细胞相比,PanCSCs比例的增加与短期生存、对化疗和放疗的耐药性、疾病复发和转移潜力增强相关。大多数化疗药物对构成肿瘤大部分的分化癌细胞有不良影响,但它们通常对PanCSCs无效。Pothula等.据报道,在治疗开始时,吉西他滨给小鼠立即导致PDAC细胞死亡,减少肿瘤体积,但对转移扩散没有任何抑制作用[210]。由于EMT的增加,吉西他滨治疗选择了一个具有更高迁移潜力的PanCSCs亚群,这可以解释吉西他滨治疗缺乏抗转移效应和转移率的矛盾增加[210]。CSCs的特点之一是药物外排泵的高表达,如多药耐药基因保护细胞免受化疗试剂的伤害[73]。此外,CSCs具有显著的修复烷基化剂和辐射引起的DNA损伤的能力[211]。此外,CSCs主要存在于细胞周期的G0期,因此对细胞周期特异性药物如5-FU具有耐药性[212]。重要的是,研究发现ALDH+ PanSCs对化疗诱导的细胞死亡具有耐药性,并且具有高度的致瘤性[213]。关于ALDH1在细胞解毒中的作用,表达高水平ALDH1的PanCSCs细胞在细胞毒性化疗下获得生存优势就不足为奇了[213]。
针对PDAC基因组不稳定性的新治疗策略目前正在研究中。种系或体细胞突变乳腺癌易感基因1而且BRCA2,自动取款机和其他参与同源重组的基因可能增加对DNA损伤细胞毒性制剂(如铂类似物)的敏感性,但也使这些肿瘤对PARP抑制剂特别敏感,使碱基切除DNA修复失效,这是一种潜在的合成致命治疗策略[214]。Veliparib是一种有效的PARP 1/2口服不可逆抑制剂,已在已知BRCA突变或有非常强的个人或家族胰腺或BRCA相关恶性肿瘤病史(这是这些突变潜在存在的指标)的患者中进行了临床试验评估[214,215]。IB期试验表明,据报道,韦利帕利布、顺铂(一种铂基药物)和吉西他滨的三联用药显示出brca相关PDAC的高活性[215]。相比之下,在非brca突变患者中没有观察到明显的活性[215],除了两种剂量限制毒性(包括中性粒细胞减少和血小板减少)外,该药的耐受性相当好,大多数是每天连续给药80mg[215]。目前,一项随机II期临床试验研究正在调查韦利帕尼联合吉西他滨和顺铂治疗已知的晚期PDAC患者的疗效,并与吉西他滨和顺铂进行比较BRCA或PALB2(ClinicalTrials.gov标识:NCT01585805)。此外,一项III期、随机、双盲、安慰剂对照、维持奥拉帕尼(parp1抑制剂)单药治疗的患者BRCA突变转移性胰腺癌,其疾病在一线铂基化疗后没有进展也在研究中(ClinicalTrials.gov Identifier: can -3716)。
目前,一项II期试验正在分析在PDAC切除术后,与单独吉西他滨相比,在吉西他滨辅助化疗中加入厄洛替尼是否能提高生存率。鉴于PDAC中直接靶向EGFR信号的复杂性,药物开发工作也聚焦于EGFR的下游成分。在这方面,靶向EGFR的下游效应因子STAT3可显著降低肿瘤负担,延缓肿瘤进展,增加对吉西他滨的反应,并减少增殖细胞[216]。此外,SRC激酶(EGFR的下游介质)抑制剂和5-FU的联合使用降低了5-FU诱导的EGFR/AKT通路的激活,并显著降低了体内肿瘤生长和远处转移[216]。
Slusarczyk最近开发了一种提高吉西他滨治疗效率的策略,即使用磷酸酰胺ProTide方法[217]et al。[217]通过在吉西他滨上添加磷酸酰胺ProTide片段,开发出Acelarin (nucu -1031),使:a)被动进入细胞;B)绕过对运输者的依赖;C)减少对磷酸化激酶的依赖,d)降低脱氨基的敏感性。Acelarin能在细胞内产生高水平的活性物质,并且患者具有良好的耐受性[217,218]。I/II期研究结果显示,Acelarin在早期化疗复发/难治性晚期实体肿瘤患者(包括吉西他滨)的疾病控制率显著达到78%[218]。特别是,一名肝转移的胰腺癌患者对Acelarin有部分反应,肿瘤体积减少30%,CEA水平减少92%,CA 19-9减少73%[218]。Acelarin在卵巢癌、胆道癌和胰腺癌的全球III期研究目前正在计划中。
Ruxolitinib是一种JAK1/JAK2抑制剂,根据生存期和症状的改善被批准用于骨髓纤维化患者,在胰腺癌小鼠模型中显示可减弱恶病质进展[219]。在最近报道的一项随机II期试验(Ruxolitinib In难治性转移性胰腺癌患者[RECAP]研究)中,在吉西他滨基础治疗进展的患者中,将Ruxolitinib加卡培他滨与卡培他滨加安慰剂进行了比较[220]。尽管在整个研究人群中没有观察到生存差异,但对c -反应蛋白(CRP)水平升高的亚组患者的预先计划分析显示,含鲁索列替尼组在统计学上有显著的生存获益[220]。c -反应蛋白(CRP)是一种特征良好且敏感的全身炎症标志物。这些有希望的结果导致了两项被称为JAK1和JAK2的注册III期试验,专门局限于具有高CRP基线水平的转移性胰腺癌患者(ClinicalTrials.gov标识:NCT02117479和NCT02119663)[220]。
考虑到代谢性疾病和PDAC的发展之间的紧密联系,在PDAC的管理中结合使用二甲双胍和降胆固醇药物已经有了一个有希望的趋势。有利的是,在PDAC确诊后使用降胆固醇药物如辛伐他汀和阿托伐他汀已被发现与非转移性PDAC患者更长的生存期相关[221]。也有一些临床证据表明,二甲双胍可以降低PDAC的发生率,改善糖尿病PDAC患者的预后[222,223]。目前提出的二甲双胍的抗肿瘤分子作用主要与抑制胰岛素/IGF1信号、下调mTOR信号、激活AMPK、中断胰岛素和GPCR系统之间的串声有关[49224,225]。然而,一项随机II期试验表明,在吉西他滨和厄洛替尼的基础上加用二甲双胍并不能改善局部晚期或转移性胰腺癌患者的预后[226]。此外,一项联合二甲双胍和紫杉醇治疗吉西他滨难治性晚期患者的II期试验未发现添加二甲双胍的任何益处[227]。
鉴于目前的化疗药物似乎在消耗CSC池方面大部分无效,它们与CSC靶向药物的联合使用可能会促进肿瘤的消退。值得期待的是,在高侵袭性原位模型中,与吉西他滨治疗相比,Notch信号通路抑制剂(gf -03084014)与吉西他滨联合使用可有效诱导凋亡、抑制肿瘤细胞增殖和血管生成,从而抑制原发肿瘤生长并控制转移扩散[228]。一种由声波HH抑制剂(环巴胺/CUR199691)、mTOR信号抑制剂(雷帕霉素)和吉西他滨组成的三联治疗能够消除患者源性胰腺肿瘤小鼠中的PanCSCs[229]。令人鼓舞的是,这种组合具有合理的耐受性,并显著延长了长期生存率[229]。因此,桑丘et al。[230]表明,由于PanCSCs强烈依赖氧化代谢,因此容易受到线粒体靶向治疗(如二甲双胍)的影响。然而,在二甲双胍治疗期间,由于其中间糖酵解/呼吸表型,最终出现了耐药克隆。桑丘et al。[230]证明了Myc的遗传/药理靶向性可以防止/恢复耐药PanCSCs对二甲双胍的反应。值得注意的是,钙通道阻滞剂维拉帕米已被证明可以通过靶向在PanCSCs中选择性过表达的ABC转运蛋白B家族成员1 (ABCB1)和ENT1的转运功能,增加化疗药物的细胞毒性作用和多药耐药[231]。
许多针对基质的策略正在研究中。其中,nab-紫杉醇由于结合了细胞毒性治疗和靶向给药,显得很有前景。nab -紫杉醇是一种胶体悬浮液,纳米颗粒均质于人血清白蛋白中,与紫杉醇结合,紫杉醇是一种微管稳定剂,可诱导细胞周期阻滞并最终导致细胞死亡[232]。SPARC结合白蛋白并促进白蛋白结合疗法向肿瘤的传递[232]。在早期临床试验中,SPARC表达较高的PDAC与接受nab-紫杉醇和吉西他滨联合治疗的患者的生存率提高有关[233]。nabb -紫杉醇还会降低PSCs的含量,导致肿瘤硬度降低[234]。同样,在小鼠PDAC模型中,使用靶向PSCs的双磷酸盐可减少纤维化、肿瘤体积/重量、腹膜播散、血管生成和细胞增殖,并增加凋亡[235]。当双磷酸盐与nab-紫杉醇联合使用时,这些体内抗肿瘤作用得到增强[235]。令人惊讶的是,虽然pscs释放的HGF抑制能有效抑制局部肿瘤生长、肿瘤血管生成和转移,但当与吉西他滨联合使用时,HGF抑制的抗转移作用消失了[236]。看来,吉西他滨增加细胞干性和迁移潜能的能力超过了HGF抑制的任何抗迁移影响[236]。 Thus, targeted therapies will require careful modelling for optimal integration with existing treatment modalities.
考虑到炎症对PDAC启动和发展的高度影响,激活特异性免疫系统成分并克服免疫逃避的方法似乎是有前途的。适当地,使用brutinib(一种Bruton酪氨酸激酶抑制剂)和使用激动剂CD40抗体反向免疫抑制和驱动T细胞依赖的抗肿瘤反应,抑制肿瘤生长和提高对标准治疗化疗的反应性[237-239]来预防B细胞活性。令人鼓舞的是,据报道,CD40激动剂抗体联合吉西他滨对转移性PDAC患者具有良好的耐受性,并与抗肿瘤活性相关[240]。同样,抑制CXCR4和堵塞CXCL12受体有望成为提高免疫检查点抑制剂(如抗ctla 4和抗pd 1抗体)疗效的药理学方法[241]。目前,plerixafor(一种CXCR4拮抗剂)持续静脉给药晚期胰腺癌患者的安全性正在调查中(ClinicalTrials.gov Identifier: am - plex, NCT02179970)。
在转移性胰腺癌的积极临床研究中,一种基于免疫的替代策略包括活减毒李斯特菌单核细胞增生疫苗载体(CRS-207;Aduro Biosciences, BERKeley, CA)[242,243]。这种细菌经过基因工程改造,包含了间皮蛋白,这是一种在大多数胰腺癌中表达的肿瘤相关抗原[241,242]。在I期试验中,该制剂被证明可诱导间皮素特异性t细胞应答,在另一项单独研究中,三名此前接受过细胞异体胰腺癌疫苗(GVAX)治疗的胰腺癌患者的生存期明显延长[242]。在此基础上,最近在化疗难治性转移性胰腺癌患者中进行了一项随机II期试验,结果显示,与GVAX/ Cy(环磷酰胺:Treg抑制剂)序次治疗相比,GVAX/ Cy对OS的改善具有统计学意义[243]。一个更大的随机研究,被称为安全性和有效性的组合李斯特菌/GVAX胰腺疫苗在胰腺癌环境(ECLIPSE)的试验,目前正在进行(ClinicalTrials-.gov标识:NCT02004262)。
最近,麦克尤恩et al。[244]已经在临床前模型中证实了声动力疗法(SDT)治疗PDAC的有效性。基本上,SDT涉及到通过刺激产生细胞毒性ROS来局部激活一种其他无毒的敏化剂[244]。鉴于PDAC是高度缺氧的,这对SDT方法的有效性产生了负面影响。因此,麦克尤恩et al。[244]旨在将氧纳入微泡(MB)的核心,以增强肿瘤微环境中产生的ROS数量,因为氧是SDT中产生ROS的基质。令人鼓舞的是,注射超声响应微泡(MB),充满了气态氧,提供了sdt介导的肿瘤显著减少。之后,麦克尤恩et al。[244]描述了氧载微泡(O2MB)平台的制备,用于使用声动力疗法(SDT)和抗代谢物疗法(5-FU)对PDAC进行高靶向治疗。本研究的目的是通过致敏微环境来提高抗代谢药物治疗的疗效,并减少化疗相关的副作用[245]。值得注意的是,结合使用O2MB偶联物的声动力和抗代谢药物治疗可增强培养的PDAC细胞系的细胞毒性,并减少异位细胞肿瘤体积[245]。这种方法是治疗局部晚期PDAC的一种有前途的替代化疗方法,可能是一种有效的新辅助治疗
近年来,研究的重点已经转向了特殊反应者,即对药物有惊人、戏剧性反应的罕见患者。美国国家癌症研究所(NCI)将异常反应定义为超过10%的患者对未预期的全身治疗发生完全或部分反应,持续至少6个月[246]。NCI在2014年9月启动了特殊反应计划,该计划试图了解癌症患者对治疗的特殊反应的分子基础,主要是通过化疗(ClinicalTrials.gov标识:NCT02243592,预计初步完成日期:2100年1月)。一份具体的报告证明了这种方法的可行性[247]。艾耶et al。247对接受mTOR抑制剂依维莫司的特殊应答者的膀胱癌组织进行了全外显子组测序(WES)分析。这种异常反应背后的生物学机制与功能缺失突变有关TSC,在该试验中,肿瘤反应良好(肿瘤收缩)的患者的肿瘤中,mTOR是一种负调控因子TSC1突变后,肿瘤没有缩小[247]。从这个角度来看,TSC1被假设为依维莫司治疗的预测反应标志物,导致了一项前瞻性试验来测试依维莫司治疗对患有TSC1-突变肿瘤[246,247]。Subbiah还报告了其他异常反应et al。[248]他证实了两名尤文氏肉瘤患者对单药IGF1R治疗有显著反应,然后复发。一旦对单药IGF1R抑制剂产生耐药性,患者接受IGF1R联合雷帕霉素治疗248在临床上,两例患者对联合治疗均有反应[248]。磷酸化的(p)-AKT和p-mTOR在耐药组织中发生上调,表明AKT/mTOR途径是获得性耐单药IGF1R治疗的机制[248]。当一名患者继续有反应时,另一名患者进展[248]。进一步的分析表明,出现的耐药肿瘤显示ERK通路的并发激活是一种潜在的耐药机制[248]。这些发现强调了这样一个事实,即癌症治疗需要一个移动的、动态的靶点规划,这可以通过对患者肿瘤组织的虚拟实时快速研究而实现。
幸运的是,在PDAC中也报告了异常反应。Chue等.【249】报道了一名转移性PDAC患者,对POLF(紫杉醇+奥沙利铂+叶酸+5-FU)和吉西他滨为基础的方案(包括奥沙利铂和叶酸/5-FU联合间歇性西妥昔单抗(EGFR抑制剂))的节律给药治疗反应非常好(从诊断到报告发表5年)。值得注意的是,病人的肿瘤显示没有喀斯特变异表明是野生型喀斯特对西妥昔单德有反应,有助于患者不同寻常的长生存期[248]等.[250]报道了一例PDAC转移到肝脏的患者,接受了吉西他滨为基础的化疗。方案包括吉西他滨和奥沙利铂(GEMOX)的周期,接着是吉西他滨、多西他赛和卡培他滨的周期,然后是吉西他滨和nabe -紫杉醇的周期,从转移性胰腺癌化疗开始2年以来,疗效异常[250]。虽然没有对该患者进行基因检测,但患者对这些治疗的异常反应可能导致该患者的肿瘤可能具有独特的基因和分子组成的结论。值得注意的是里奥斯·佩雷斯等.[251]报道了一例50岁女性原发性PDAC合并两处肝转移和ca19 -9水平高的病例。患者在3个月内接受6个周期FOLFIRONIX治疗,最后一个月停用奥沙利铂[251]。观察到CA 19-9的显著减少和两种肝转移的完全消退[251]。由于没有证据表明存在远处疾病,我们对正常的CA19-9进行了以卡培他滨为基础的同步全身和局部化疗8个月[251]。ca19 -9的轻度增加促进了重新分化,表明原发肿瘤活跃,但没有远处的疾病[251]。然后,对该患者进行惠普尔检查,使其未发现可检测的癌症[251]。切除时从最初的IV期降至IIB期,迄今已有28个月的存活时间[251]。在体外化学敏感性试验中,对患者的肿瘤组织进行了治疗方案敏感性的检查,结果表明,与吉西他滨相比,该患者的肿瘤样本对FOLFIRINOX非常敏感。该检测方法正在开发中,作为一项临床试验的一部分,该试验将前瞻性地测试为个别患者确定最有效治疗方法的能力[251]。
尽管这种方法很有前景,但也存在一些挑战。首先,对于每种被描述的异常反应机制,重要的是要证明发生异常反应的肿瘤确实依赖于这种改变,并且描述这种改变的功能意义是至关重要的。其次,通常很难检测肿瘤对特定改变的依赖性,也很难找到一种药物或多种药物的组合,专门针对特定的改变。因此,患者驱动细胞系或患者源性异种移植的产生可能最终使与特定基因组和表型改变相关的依赖关系的更明确的评估具有对抗癌药物的特殊反应的潜力。同时,特殊的应答病例也可能促进治疗耐药的分子机制的识别。此外,设计具有特殊应答者的临床试验将允许更快地发现癌症生物学的途径,并开发新制剂或指导现有癌症疗法的使用。重要的是,这种方法将有助于预测抗癌药物的主要临床反应。
目前,有一项针对转移性胰腺癌患者的观察性研究正在进行中,以确定对标准治疗化疗异常应答者和无应答者之间的基因组差异(ClinicalTrials.gov Identifier: NCT02555735)。
这些例子突出了个体化医疗的重要性,它指导所有肿瘤的分子分析,并帮助确定最合适的联合疗法。
如前所述,目前PDAC的临床管理主要依赖于标准方案。然而,由于肿瘤之间的异质性,以及影响药物代谢、药物运输、个体对药物的敏感性和患者的表观遗传背景的患者之间的基因型差异,对所有患者使用同一治疗方法将导致变化和不可预测的反应。因此,个性化医疗(PM)为pdac治疗和护理提供了一种有吸引力的方法。
PM可被定义为“一种利用个人基因、蛋白质和环境信息来预防、诊断和治疗疾病的药物”(美国国家癌症研究所2011年)。PM实现了“组学”科学(基因组学、表观基因组学、转录组学、蛋白质组学、代谢组学等),以整合各种数据集,目的是解剖分子特征和功能途径,帮助对肿瘤亚型进行分类,并确定其自然过程、预后和对治疗的响应性。然而,为了确保真正个性化的方法,必须考虑时间上的遗传和表观遗传异质性、微生物区系和广泛的环境因素,包括营养、压力和调节疾病和对治疗反应的其他因素。在这方面,无论是预防还是PDAC的治疗管理,个性化的生活方式药物都可以整合到个人的整个生活中。
理想情况下,这应该包括使用诊断性生物标志物的非侵入性筛查方案的早期检测策略,包括患者和二级保健医生及其支持人员在预后性生物标志物的指导下进行合作决策,提供个性化的多因素治疗,并在预测性生物标志物指导下进行监测方案。以及使用生物标志物和生活质量标志物对患者预后进行持续监测(表6、图5)。
表6.个体化药物在PDAC管理中的潜在应用。
目标 |
方法 |
整体基本原理/评论 |
|
诊断 |
具有高特异性和敏感性的最小/非侵入性多重生物标志物方法 |
多种生物标志物方法提高早期检测和监测的有效性。 |
诊断BM面板 |
诊断样本 |
灵敏度(%)(HCvsPDAC) |
特异性(%)(HCvsPDAC) |
3-蛋白板(TNC/TFPI/CA19-9) [303] |
等离子体 |
97 |
90 |
3蛋白板(ca19 -9, ICAM1, OPG) [304] |
血清 |
78 |
94 |
3蛋白板(ca19 -9, CEA, TIMP1) [304] |
血清 |
71 |
89 |
三蛋白组(Lyve1, reg1a, tff1) [305] |
尿液 |
77 |
90 |
miRNA面板(-21,-210,-155,-196a) [306] |
等离子体 |
64 |
89 |
5-CpG网站(IL10_P348,LCN2_P86,ZAP70_P220,AIM2_P62 TAL1_P817)[307] |
循环白细胞 |
65 |
90 |
25个细胞因子+补体面板(C1酯酶抑制剂、C3、C5、CD40、Eotaxin、GM-CSF、IgM、IL-11、IL-12、IL-16、IL-1α、il -1 ra、IL-2、IL-3、IL-4、IL-7、整合素α-10、MCP-1、-3、Mucin-1、Properdin、TGF-α、TGF-β1、TNF-β、VEGF) [308] |
血清 |
73 * *非PDAC (HC, AIP, CP) vs PDAC |
75 * *非PDAC (HC, AIP, CP) vs PDAC |
自身抗体面板(抗ctdsp1, -MAPK9和-NR2E3 IgG) [309] |
血清(自身抗体水平高于对照组) |
太喀斯特,太TP53,太SMAD4,异常DNA甲基化7基因面板(FOXE1,NPTX2,CLDN5,CDKN2A,TFPI2,SPARC, ppENK) [20] |
家族性和散发性PDAC组织。(用于筛查早期非侵入性肿瘤)。 |
|
治疗 |
潜在的个性化综合治疗策略 |
|
药物及其作用机制 |
药物的预测BM和临床影响 |
预后BM |
可能针对BM的天然化合物 |
天然化合物和化疗药物的组合治疗,以促进治疗的协同作用,并最终提高总体疗效 |
- 吉西他滨:阻止DNA合成
- 吉西他滨+击倒STAT3:增加对吉西他滨的反应[216]
- 吉西他滨+HGF抑制:降低茎干和迁移[236]
|
- 的单核苷酸多态性CMPK1,CDA, dCK, RRM1, RRM2, ENT1, SLC29A1与吉西他滨反应相关[310-313]
- ↓ENT1,↓CNT1,↓CNT3,↓dCK↓TK2,↑RRM1,↑RRM2表达与吉西他滨耐药相关[174-177]。
- ↓p53,↓PTEN,↑Bcl家族蛋白,↑NF - κB和↑Hif-1α,↑SRC,↑EGFR,↑STAT3,↑PI3K/AKT,↑Notch,↑MAPK,↑HGF pw与吉西他滨耐药相关[178-184,236]
- ↑miR-21,↓miR-200,↓let-7与吉西他滨耐药相关[180,289]
|
- 的单核苷酸多态性CMPK1,CDA dCK RRM1ENT1 RRM2,SLC29A1与3/4级中性粒细胞减少症和PFS相关[310-312]
- ↑miR-21,↓miR-200与PDAC细胞的侵袭性相关[180,289]
|
- 姜黄素及其类似物,辣椒素,类黄酮,异硫氰酸酯,白藜芦醇。
|
- 姜黄素或辣椒素、黄酮类、异硫氰酸酯或白藜芦醇联合吉西他滨可逆转吉西他滨耐药性,抑制增殖、侵袭、血管生成和转移,增加细胞凋亡[180,289,314-317]
|
- 研究者用:抑制TS、RNA和DNA合成
- PP2: SRC激酶抑制剂
- 研究者用+ PP2:降低5- fu诱导的EGFR/AKT活化pw。
|
- H3K4me2, H3K9me2, H3K18ac修饰与吉西他滨反应相关[258]
- ↑TS,↑DPD,↓CNT1,↑MRPs与5-FU耐药相关[179,187,188,318]
- EGFR/AKT/SRC通路与5-FU耐药相关[185]。
|
- ↓H3K4me2,↓H3K9me2,↓H3K18ac均与低生存率相关[258]
- ↑DPD、↑TS、↑p-Src均与生存率差相关[185]
|
- 黄酮,白藜芦醇,辣椒素,异硫氰酸盐,姜黄素,叶酸,叶酸,维生素B6B12和蛋氨酸。
|
- 黄酮+5-FU处理抑制细胞增殖,诱导细胞凋亡[294]
- 姜黄素或类黄酮或异硫氰酸酯作为组蛋白去乙酰化酶抑制剂,可能与5-FU协同作用[258,315,316]
- 叶酸通过抑制TS增强5-FU的作用,从而显著和长期抑制DNA合成[320]
- 姜黄素或辣椒素或黄酮类或异硫氰酸酯或白藜芦醇下调EGFR、AKT、SRC信号通路可能增加5-FU的抗肿瘤活性[315-316]
|
|
- 以铂为基础的代理(如奥沙利铂、顺铂)损伤DNA。
- Veliparic, olaparib: PARP抑制剂
- 铂剂+ PARP抑制剂:
代表了一种合成致死性治疗策略 |
- 的不足。RADs, ATR, ATM, CHK1, CHK2, BRCA1, BRCA2, FANCG, FANCCPALB2, ERCC1与铂类药物和PARP抑制剂敏化反应的改善相关[60,215,321]
- ↑PER2与改善顺铂反应相关[301]
|
|
|
- 异硫氰酸酯作为补充PARP治疗的可能治疗机制[295]
- 黄酮联合顺铂诱导细胞凋亡[303]
|
|
- FOLFIRINOX:(5-FU,叶酸,伊立替康,奥沙利铂)
- 伊立替康:拓扑异构酶1抑制剂
- 叶酸:叶酸酸
|
- FOLFIRINOX对ERCC1水平正常的患者比ERCC1高表达患者更有效[321]
|
|
|
|
|
|
- EGFR/MET, EGFR/IGFR, EGFR, erbB-2/EGFR异质二聚体↓PTEN,↑AKT与厄洛替尼耐药相关[179,195,196]
- wtKRAS、↑EGFR与厄洛替尼疗效相关[323]
|
- wtKRAS与厄洛替尼治疗PDAC的改善OS相关[323]
- mtKRAS与[23]生存率降低相关
|
|
- 姜黄素、辣椒素、黄酮类、异硫氰酸酯、白藜芦醇下调EGFR、AKT、SRC信号通路可能增加厄洛替尼的抗肿瘤活性[315,316]
|
|
|
- SPARC促进了nabb -紫杉醇向肿瘤的传递[324]
|
- 瘤周间质中的SPARC与不良预后相关[324]
- SPARC与接受nab-紫杉醇和吉西他滨联合治疗的患者生存率提高有关[324]
|
|
- 维甲酸或vit A或vit D3的使用可能会降低PSCs的活性,以协同nab-pactitaxel的作用并降低肿瘤硬度[234,315,316]。
|
|
个性化的生活方式推荐 |
饮食干预 |
体育活动干预 |
身心干预 |
环境干预措施 |
综合肿瘤学的应用,以达到最佳的结果和最大生命质量 |
- 热量限制[280]
- 生酮饮食[286]
- 限制食物进入暗期(活性期8-9小时)[302]
- 饮食包括大量食用新鲜水果和新鲜蔬菜、维生素C、维生素E、omega-3脂肪酸,少量食用红肉、饱和脂肪、加工食品、添加糖、软饮料和加糖水果汤或炖水果[272]
|
|
- 冥想,[269270]
- 定期锻炼[274]
- 瑜伽[274]
|
- 每天只喝一杯酒[272]
- 戒烟,
- 避免吸烟;传染性病原体;辐射;工业化学品、污染和药物。
|
- 时间限制喂养可改善糖耐量和营养稳态,减少胰岛素抵抗、全脂堆积和炎症。
- 健康的饮食
- 增加身体活动,
- 保持健康的体重
- 避免外部和内部毒物
- 合理的昼夜/睡眠周期挽救生物钟,保持新陈代谢健康
- 放松策略,以减少压力,改善情绪,改变健康行为,坚持癌症治疗和减少吸烟。
|
(BM:生物标志物:Mt:突变;wt:野生型;HC:健康对照组;PDAC;胰导管腺癌;AIP:自身免疫性胰腺炎;CP:慢性胰腺炎CA 19-9:癌抗原19-9;TNC:腱蛋白C;TFPI:组织因子通路抑制剂;ICAM1:细胞间粘附素分子1; OPN: osteopontin; CEA: carcinoembryonic antigen; TIMP-1: metalloproteinase inhibitor 1; LYVE1: lymphatic vessel endothelial hyalunoric acid receptor 1; REG1A: lithoastathine-1-alpha; TFF1: trefoil factor 1; C1 esterase inh.: C3: complement 3; C5: complement 5: CD40: cluster of differentiation 40; GM-CSF: granulocyte macrophage colony-stimulating factor; IgM: immunoglobulin M; IgG: immunoglobulin G; IL-11: interleukin-11; IL-12: interleukin-12; IL-16: interleukin-16; IL-1α: interleukin-1 apha; IL-1-ra: interleukin 1 receptor antagonist: IL-2: interleukin-2; IL-3: interleukin-3; IL-4: interleukin-4; IL-7: interleukin-7; Integrin α-10: integrin alpha-10; MCP-1: monocyte chemoattractant protein-1; MCP-3: monocyte chemoattractant protein-3; TGF-α: transforming growth factor alpha; TGF-β1: transforming growth factor beta-1: TNF-β: tumor necrosis factor alpha; VEGF: vascular endothelial growth factor; CTDSP1: carboxy-terminal domain: RNA polymerase II: polypeptide A small phosphatse 1; MAPK9: mitogen-activated protein kinase 9; NR2E3: nuclear receptor subfamily 2 group E member 3; SNP: single nucleotide polymorphism; CMPK1: cytidine/uridine monophosphate kinase 1; CDA: cytidine deaminase; dCK : deoxycytidine kinase; RPM1: ribonucleotide reductase 1; RPM2: ribonucleotide reductase 2; ↑: increase; ↓: decrease; ENT: equilibrative nucleoside transporter ; SLC29A: slute carrier family; STAT3: signal transducer and activator of transcription 3; HGF: hepatocyte growth factor; pw: pathway; MAPK: mitogen activated kinase: NF–kB: nuclear factor-kappa-B; PTEN: phosphatase and tension homolog; AKT2: v-AKT thymoma viral oncogene homolog 2; PI3K: phosphoinositide-3-kinase; Hif-1α: hypoxia-inducible factor 1-alpha; H: histone; me2: dimethylation; ac: acetylation; K: lysine; R: arginine; PFS: progression free survival;研究者用:氟尿嘧啶;胸苷酸合成酶;表皮生长因子受体;上皮生长因子受体;CNT1:核苷转运蛋白;MRPs:多重耐药转运蛋白;DPD:二氢嘧啶脱氢酶:Vit:维生素;DNA:脱氧核糖核酸;例如:例子;PARP:聚(adp -核糖)聚合酶; ATR: ATR serine/threonine kinase; ATM: ATM serine/threonine kinase; CHK1: checkpoint kinase 1; CHK2: checkpoint kinase 2; BRCA1: breast cancer type 1 susceptibility protein; BRCA2: breast cancer type 2 susceptibility protein; FANCG: fanconi anemia complementation group G; FANCC: fanconi anemia complementation group C; PALB2: partner and localizer of BRCA1/2; ERCC1: excision repair cross-complementation group 1; PER2: period circadian clock 2; SPARC: secreted protein acidic and cysteine rich; PSC: pancreatic stellatte cell; p-SRC: phospho-SRC; OS: overall survival; QOL: quality of life; ↓: low expression; ↑: high expression

图5。生物标志物在PDAC治疗中个性化综合治疗方法的潜在应用示意图。将遗传和功能数据结合到网络建模中,预测表型PDAC多样性,为个体选择合适的治疗方法,并整合最佳的循证和临床验证的补充方法,满足个体减少症状和改善生活质量的需求。
(缩写:BM,生物标志物;和,与;Wt,野生型;异烟肼,抑制;Cyt +comp信号,细胞因子,互补信号;NLR:中性粒细胞与淋巴细胞比值;喀斯特;柯尔斯滕大鼠肉瘤病毒癌基因同源物;宝石,吉西他滨;CDA:胞苷脱氨酶; CMPK1, cytidine monophosphate (UMP-CMP) kinase 1; DPD, dihydropyrimidine dehydrogenase; CDF, curcumin analogue; SNPs, single nucleotide polymorphisms; 5’-FU, fluorouracil; QOL, quality of life; dCK,脱氧胞苷激酶;HR,同源重组;NC,天然化合物;↓,低;↑,高)
通过筛查高危人群,可及时发现具有准确的早期疾病非侵入性生物标志物的PDAC。特别是,开发“多重生物标志物面板”,以提高筛查试验监测风险个体的敏感性,被认为有很大潜力提高早期检测的诊断准确性。选择筛查人群的实用方法可以基于流行病学证据和临床参数,包括肥胖、糖尿病、慢性胰腺炎或遗传性PDAC综合征。除了个人、家庭和遗传史,环境因素也应被考虑到风险分层和制定量身定制的筛查和监测计划。例如,考虑到糖尿病和PDAC之间的联系,选择代谢相关基因中的SNPs如IGF1,ADIPOQ可提供一种重要的筛查工具,帮助识别罹患PDAC风险增加的个体。
显然,对耐药机制的先验知识的要求,在避免过度毒性的情况下早期开始抗癌治疗,将预测生物标志物带到癌症治疗的中心。然而,由于pdac之间可用的组织较少和异质性,为计划治疗和监测策略确定预后和预测性生物标志物一直具有挑战性。事实上,需要在疾病的不同阶段获得转移性组织和组织样本,以及在治疗过程中不断变化的肿瘤生物学景观和不断生长的耐药克隆,阻碍了有效治疗的发展。因此,循环生物标记物的使用似乎更可取,因为它们在疾病和治疗过程中易于收集,而且性质上相对无创。其中,自身抗体、循环肿瘤细胞(CTCs)、肿瘤释放蛋白、-代谢产物和肿瘤衍生的细胞外囊泡(EVs)在过去几年得到了重视。重要的是,在有症状的疾病出现前数月到数年,自身抗体的存在为开发有用的诊断和预后工具带来了希望。值得注意的是,除了作为一种潜在的生物标志物外,ctc还有望在患者的整个临床过程中对PDAC细胞的肿瘤遗传学、蛋白质组学和分子生物学以及药效学进行反复研究。已获得进入循环系统能力的CTCs已在40%-100% PDAC患者的腹膜液和外周血中检测到[252,253]。CTCs的异质性群体允许表型鉴定用于治疗分层[252]。由于CTCs显示肿瘤启动能力,有助于远处转移,检测CTCs可能增加早期发现转移的可能性。 This may improve prognosis following surgical resection by identifying patients who are appropriate candidates for early treatment with systemic therapy [252,253]. In addition, preoperative neutrophil-to-lymphocyte ratio (NLR) has been proposed a promising predictor of survival in patients with PDAC [254]. Remarkably, a novel, non-invasive three-protein biomarker panel that is able to detect patients with early-stage PDAC completely non-invasively, through analysis of urine samples have been established [255]. Sampling pancreatic juice or tumour during EUS could be of great clinical value providing biomarkers [256]. Strikingly, Stratfordet al。[257]报道了一种6个基因的特征,可以根据患者的存活情况区分高风险(侵袭性)和低风险(侵袭性较低)肿瘤。当研究人员对一组来自局部切除PDAC患者的独立肿瘤样本使用这种方法时,他们发现高危肿瘤患者的平均生存时间为15个月;55%的患者术后存活了一年,剩下的低风险肿瘤患者平均存活时间为49个月;91%的患者在术后1年仍存活[257]。关于Whipple手术的风险(2%-6%的患者在该手术中死亡,超过50%的患者有严重的术后并发症),该特征的预测能力可能用于帮助临床医生和患者对他们的治疗计划做出决定[257]。组蛋白分析也在研究中,以绘制PDAC的组蛋白修饰模式。Manuyakorn的III期临床研究等.[258]研究表明,组蛋白修饰的细胞水平定义了先前未被识别的PDAC患者亚群,这些亚群具有不同的表观遗传表型和临床结果,并代表预后和预测性生物标志物,可为临床决策提供信息,包括使用5'FU化疗[258]。此外,可在外周血、胸腔积液和尿液中检测到的EVs可用于检测和分析肿瘤起源的分子货物,如miRNAs、突变基因组片段、脂类、蛋白质以及监测疾病的长期进展[259]。例如,Melo等人报道了PDAC患者中glypical的表达增加。此外,他们发现在PDAC小鼠模型中PanIN病变形成之前也检测到草甘酸阳性ev,随着时间的推移比例增加。此外,在glypican-1阳性ev中检测突变Kras转录本突出了它们在识别癌症特异性遗传缺陷方面的潜在用途[260]。
最近,研究集中在选择和验证方法,根据分子特征对肿瘤进行分层,并允许识别不可能从现有化疗中获得临床益处的患者。加扎利说,等.[261]报道了基于rna的检测方法,即rnascope技术,可以可靠地检测生物标志物ENT1、CDA、dCK,这些标志物被认为与PDAC细胞对吉西他滨的耐药有关[308]。目前,一项临床试验正在调查中,旨在发现可能的生物标记物,以预测Acelarate比吉西他滨的额外益处,用于后续验证(Acelarate, ISRCNT 16765355)。在一项回顾性研究中,Orlandiet al。[262]。比较了吉西他滨与FOLFRINOX在有ENT1评价的转移性PDAC患者中的疗效。无论ENT1表达如何,与吉西他滨相比,FOLFIRINOX治疗的OS和PFS在统计学上更长[262]。然而,当根据ENT1表达对患者进行分层时,无论是FOLFIRINOX还是吉西他滨治疗的ENT1阳性患者,OS均未发现差异[262]。值得注意的是,与吉西他滨治疗的ENT1阴性患者相比,吉西他滨治疗的ENT1阳性患者的OS和PFS均有显著的统计学改善[262]。因此,本研究证明了ENT1在预测吉西他滨活性方面的有效性,并为ENT1表达作为吉西他滨的预测因子提供了证据[262]。由于吉西他滨比FOLFIRINOX有更好的安全性,它可能更适合ENT1阳性的晚期疾病和性能较差的患者。回顾基于铂的药物在brca相关癌症中的疗效,PDAC中的基于铂的治疗应该针对含有HR通路相关基因突变的肿瘤患者。
许多分子靶向疗法,以特定的生物标记物为指导,已经出现在癌症化疗中,并显示出相当大的前景。例如,过表达EGFR的肿瘤受益于EGFR靶向剂[263]。值得注意的是,如果已经存在匹配的治疗方法,并且可以“重新利用”或“挽救”生物标志物,那么累积起来,它们提供了一个潜在的重要机会,可以在比新的治疗方法的发现和开发更短的时间内改善结果。考虑到这一点,EGFR靶向治疗可以挽救野生型PDAC患者的肿瘤喀斯特和过表达EGFR。显然,在一项随机、开放标签、前瞻性试验中,吉西他滨联合厄洛替尼辅助化疗被发现比单独吉西他滨治疗转移性PDAC更有效,尤其是EGFR突变患者(ClinicalTrials.gov编号,NCT01608841)[264]。
考虑到TSC1突变患者使用mTOR抑制剂的异常反应,在PDAC患者的亚人群中评估mTOR抑制剂的重新使用可能是有价值的,这些PDAC患者的肿瘤涉及基因的基因改变LKB1,TSC1,PTEN.
显然,根据生物标记物确定肿瘤亚型提供了测试靶向药物在PDAC患者中用于其他类型癌症的疗效的机会,这些肿瘤包含相同的分子靶点。然而,这一策略还不成熟,在应用时需要格外小心。为了解决这个问题,原代细胞培养模型似乎是可测试的,可以准确定义肿瘤的分子图谱并预测治疗反应。令人鼓舞的是,戈兰高地et al。[265]开发了一种独特的腹水来源的PDAC原代细胞培养模型,以研究PDAC进展中的信号通路,并评估针对个别患者的靶向治疗。此外,张et al。[266]已经成功地从患者活检组织中培养出PDAC类器官用于药物测试,旨在为这种恶性肿瘤的建模和治疗提供新的个性化方法。
令人遗憾的是,由于遗传易感性、以前的生活方式、心理/身体创伤、未来的生活方式和抗肿瘤治疗,癌症幸存者患其他疾病和继发性癌症的风险更大。此外,癌症及其治疗导致的功能状态下降和残疾增加极大地影响了癌症幸存者的生活质量。研究表明,基于证据的补充方法与标准医学治疗结合使用可能有助于促进肿瘤积极治疗和存活率[267,268]。补充疗法包括按摩疗法、针灸、身心疗法、音乐、疗法、体育锻炼、营养和营养补充以及其他疗法[269,270]综合肿瘤学旨在最佳地结合传统疗法和最佳补充疗法,以积极影响预后并提高生活质量,无论患者是否接近生命的尽头[265-268]。随着个性化癌症护理的发展和越来越多的证据支持多因素综合疗法的有效性,个性化治疗方案可能比传统治疗更有可能满足患者的需求,同时也有助于提高癌症患者的生存和生活质量。生物标志物在临床实践中的应用不仅可以促进个性化治疗方案的设计和更好地预测临床结果,还可以提供提高生存率和生活质量的信息。可被身体活动、饮食、压力和环境因素改变的生物标记物可能有助于规划个性化的生活方式和监测对干预措施的反应。例如,观察到的体育活动、超重或肥胖和癌症之间关联的相关生物标志物是性类固醇激素、高胰岛素血症和胰岛素抵抗、代谢激素、炎症增加、免疫功能下降和氧化应激[271]。
因为中枢性肥胖和肥胖是已知的PDAC危险因素272]。运动通过减少胰岛素抵抗和胰岛素/IGF1分泌对肿瘤预后有潜在的有益影响。此外,脂肪的减少会导致脂肪细胞产生的促炎因子(如tnf - α、IL-6、瘦素)的减少[273]。最近的一项随机对照试验显示,受疲劳影响严重的晚期PDAC患者很可能从运动干预中受益[274]。然而,由于多种PDAC相关症状,如疲劳、抑郁、疼痛和营养不良,运动对患者来说可能具有挑战性。因此,除了常规治疗外,国际适应身体活动联合会(APA)还建议晚期PDAC患者进行适应身体活动[274]。APA计划的实施包括根据患者的身体健康、运动类型偏好、心理功能和期望进行个体化活动)、癌症(分期、治疗和耐受性)和社会环境[274]。
PDAC患者经常经历严重的疼痛、疲劳、焦虑和抑郁以及恐惧和压力,导致生活质量受损,治疗依从性降低,生存期降低[274,275]。特别是,长期的情绪困扰通过下调NK活性导致免疫系统的抑制或失调,而这在PDAC中是不存在的[275]。此外,压力可能是PDAC启动和进展的辅助因素,因为儿茶酚胺应激激素(去甲肾上腺素)也因吸烟而升高,被证明可以诱导PDAC细胞和不朽导管细胞的自我更新和生长[276-278]放松与通过调节细胞因子和其他机制减少应激诱导的心理或生理反应有关[277,278]。因此,放松策略,如冥想、锻炼、瑜伽、其他身心干预措施可以减轻压力、改善情绪、改变健康行为、坚持癌症治疗和减少吸烟。
大量证据表明,过度氧化应激引起的细胞损伤是诱导胰腺炎症导致遗传毒性和癌症的主要机制。因此,尽量减少对外源生物的暴露可能对PDAC有保护作用。简单地说,与PDAC关联最大的证据是烟草烟雾[10-12]。尚无确凿证据表明PDAC的发展与以下因素有关:传染因子、辐射、某些工业化学品、空气污染物、一些食品和一些药物[10,11]。炎症、脂质过氧化、氧化应激、疾病状态、感染和菌群等过程产生的代谢物可能被认为是一种内部环境因素,可能与PDAC风险有关[10,11]。
众所周知,富含水果和蔬菜的饮食可以降低PDAC的风险[272]。因此,一项基于人群的大型病例对照研究显示,摄入更多的omega-3脂肪酸、维生素C和E可以降低PDAC的风险[272]。在各种研究中,乳制品被认为可以预防和促进PDAC。然而,最近一项14项队列研究的综合分析发现,成年期饮食食物、钙和总维生素D摄入量与PDAC风险无相关性[279]。大量食用红肉、饱和脂肪、某些单不饱和脂肪酸和加工食品可导致或加速PDAC[10,272]。WCRF指出,红肉是铁的来源,可以导致自由基的产生[272]。在高温下烹饪时,红肉还可能含有杂环胺和多环芳烃[272]。此外,如果饮酒,建议女性每天不超过1杯,男性每天不超过1杯[272]。关于糖代谢受损在PDAC发病过程中的作用,建议通过控制总热量摄入和增加身体活动来调节能量平衡,以控制体重,以预防或作为PDAC的治疗部分[272]。特别是,热量限制(CR)和ketegonic饮食(KD)已成为治疗癌症的有效手段。CR是一种慢性养生法,其目标是在不限制必要维生素和营养的情况下,将总热量摄入降低到比典型饮食低20-40%的水平。 reduction in total energy intake but isonutrient vitamins, minerals, fatty acids, and amino acids relative to an ad libitum-fed control regimen [280]. CR, which prevents or reverse obesity, improves insulin sensitivity, and inhibits the development and progression of a variety of cancer including PDAC [280,281]. CR regimen in PDAC decreases circulating levels of leptin and IGF-1, reduces expression of Glut1, suppresses activation of Akt/mTOR, ERK, STAT3, NF–κB and activates AMPK and SIRT1 [281-283] KD, is described as a high-fat and low carbohydrate diet that elevates circulating levels of ketone bodies, serving as an alternative energy source [284] Shuklaet al。[284]发现酮体逆转PDAC细胞的代谢适应,诱导生长停滞和凋亡。酮体处理可降低PDAC细胞中的葡萄糖摄取、糖酵解通量、谷氨酰胺摄取、乳酸分泌和ATP含量重要的是,在PDAC的细胞系模型和动物模型中,酮体对肿瘤细胞的代谢重编程是减少癌细胞诱导恶病质的原因。此外,考虑到炎症和代谢改变的重要作用,生酮饮食也提供了一种有效的治疗策略,因为通过生酮饮食降低乳酸产量已被证明可降低MDSC频率,从而提高抗肿瘤免疫反应值得注意的是,生酮饮食的副作用最小,正如先前证明的那样,2- 7mm的酮体浓度可以达到而不引起临床酸中毒回顾事实,在诊断时,约80%的PDAC患者存在恶病质,生酮饮食可能作为抗恶病质剂和抗癌剂。284-286
考虑到目前常规化疗的局限性,包括严重的毒性,化疗耐药的发展和癌症患者生活质量下降,开发安全、有效、已知和可预测的作用机制的替代药物是非常必要的。事实上,克服PDAC细胞的化疗耐药可以显著延长患者的生存期。在这方面,以优化癌细胞细胞毒性同时最小化全身毒性为目标的联合治疗似乎是PDAC最合理的初级临床管理策略。不幸的是,大多数研究倾向于实验室或动物模型,而不是基于临床。这在一定程度上是因为转移性PDAC和性能状况不佳的患者通常被排除在新的全身治疗的临床试验之外,因为担心这些患者可能无法耐受联合化疗方案带来的更大毒性。因此,联合治疗的使用促使研究使用饮食中丰富的天然化合物作为增效剂,以加剧PDAC的药物细胞毒性。近年来,越来越多的证据表明,许多天然化合物可以通过靶向遗传和/或表观遗传机制和调节信号通路,有效地逆转、抑制或阻止癌症的起始、促进或进展。因此,某些饮食中含有的天然植物性化合物具有预防和抑制癌症的潜在能力,因此有望成为化学预防/治疗干预手段(表5)。
表5.研究与PDAC相关的天然产物及其各自的作用机制[315-317]
化合物(s) |
自然源 |
行动方式 |
主要目标 |
姜黄素及其类似物 |
姜黄(姜黄) |
↓炎症,↓增殖,↓侵袭,↓存活,↓克隆原性,↑凋亡 |
↓NF-κB pw,↓IL-8,↓IL-8 pw,↓COX-2,↓STAT3,↓Notch-1 pw,↓PI3K/AKT/mTOR pw,↓EGFR,↓MAPK pw,↑ATM/CHK1 pw,↑TNFR pw,↑caspase -8,3,↑PTEN,↓前列腺素E2,↓miR-21,↑miR-200,↓HDACs,↓DNMT1 |
辣椒素 |
辣椒 |
↓炎症,↓疼痛,↓增殖,↑细胞凋亡,↑ROS生成 |
↓Bcl-2,↓PI3K/AKT pw,↓MAPK pw,↓Hedgehog pw,↓NFκB pw,↑MKK4 pw,↑JNK pw,↑caspase-3,9 |
类黄酮 Epigallocatechin-3-gallate,山柰酚 |
水果,蔬菜,树叶,谷物 茶树 (绿茶) |
↓炎症,↓增殖,↓EMT,↓侵袭,↓迁移,↓转移,↓自我更新,↓克隆性, ↓血管生成,↓厌氧糖酵解,↓葡萄糖消耗,↓脂肪生成,↑细胞凋亡, ↑ROS生成 |
↓NFκB pw,↓JAK/STAT3 pw,↓EGFR pw,↓KRas/B-raf/MAPK pw,↓PI3K/Akt/mTOR pw,↓Notch pw,↓Hedgehog pw, c-↓Myc,↓Nanog,↓Oct-4,↑c- jun,↓MMP-2,7,9,12↓VEGF,↓IL-1,↓IL-8,↓IL-6,↑caspase-8,3,↑p21 pw,↓JNK pw,↓LDHA,↓AMPK pw,↓HDACs,↓DNMT pw,↑p53 |
异硫氰酸酯 |
十字花科蔬菜 |
↓增殖, ↓自我更新,↓血管生成,↑细胞凋亡, ↑基因组稳定性 |
↓Hedgehog pw,↓NF-kappa-B pw,↓AKT pw,↓MAPK pw,↓STAT3 pw,↓HDAC pw,↓MMP-2,↓VEGF,↓VEGFR pw,↓Hif-1α,↓STAT3 pw,↓IL-6,↑PARP,↓HDAC1 |
白藜芦醇 |
红葡萄,花生,浆果,黑巧克力,日本虎杖 |
↓增殖,↓存活,↑凋亡,↓EMT,↓多能性, ↓迁移,↓入侵, ↓生长+↑酸性环境中的细胞凋亡 |
↓刺猬pw, MMP-2,9,↓PI3K/Akt/NFkB pw,↑p21,↓Src pw,↓STAT-3,↑caspase-3/7,↓Bcl-2,↓Zeb-1,↓蛞蝓,↓蜗牛,↓Nanog,↓c-Myc,↓Sox-2,↓10 -4,↓ABCG2, |
叶酸,维生素B6B12和蛋氨酸 |
全谷物,绿叶蔬菜,橙子和豆类 |
↑DNA完整性,↑DNA修复,↑DNA甲基化 |
↑DNMTs, ↑S-adenosylmethionine |
维甲酸和维生素A |
牛肝(丰富的),水果(如杏,芒果,橙子,西瓜,黑莓,桃子),蔬菜(如胡萝卜,南瓜,羽衣甘蓝,菠菜,红薯),谷物 |
↓增殖,↓迁移,↓EMT,↓纤维化,↑凋亡 |
↓PSCs活性,↓ECM,↓IL-6,↓Wnt pw |
维生素D3 |
鱼肝油(丰富)、鱼油、乳制品 |
↓增殖,↓纤维化,↑凋亡 |
↓PSCs活性,↓ECM |
IL:白介素;NF-kB:核因子-kappa- b;COX-2:环氧合酶2;AKT2: v-AKT胸腺瘤病毒癌基因同源物2;PI3K: phosphoinositide-3-kinase;STAT3:转录信号换能器和激活器3;MAPK,丝裂原激活蛋白激酶;PTEN:磷酸酶和张力同源物;Hif-1α:缺氧诱导因子1- α;EGFR:上皮生长因子受体; HDACs: histone deacetylases, DNMT: DNA methyltransferase; pw: pathway; JAK: janus kinase ATM serine/threonine kinase; CHK1: checkpoint kinase 1; MKK4: mitogen-activated protein kinase kinase 4; VEGF: vascular endothelial growth factor; TNF: tumor necrosis factor; MMP: matrix metalloproteinases, AMPK: AMP-activated protein kinase; PARP: poly(ADP-ribose) polymerase; SOX-2: SRY-Box 2; ECM: Extracellular matrix; EMT: epithelial mesenchymal transition; ABCG2: ATP binding cassette subfamily G member 2; PSC: pancreatic stellatte cell; ↓: low expression; ↑: high expression; e.g: example, ROS: reactive oxygen species.
大约十年来,吉西他滨单一疗法一直是治疗表现不佳患者的金标准。因此,人们对使用有潜力增强吉西他滨治疗潜力的天然产品很感兴趣。体外研究表明,表没食子儿茶素-3-没食子酸酯(EGCG)通过激活caspase-3和PARP,抑制JAK/STAT3信号通路,增强吉西他滨诱导的细胞凋亡[287]Aliet al。[180]表明,二氟化姜黄素(CDF),一种姜黄素类似物,通过增加miR-200的表达和降低miR-21的表达,逆转吉西他滨耐药,从而导致PTEN的重新激活。他们进一步证明,CDF可以通过失活NF-κB和COX-2使PDAC细胞对吉西他滨敏感[180]。可喜的是,一项I/II期研究显示,每日口服8g姜黄素与吉西他滨为基础的化疗联合治疗胰腺癌患者是安全可行的,但其疗效尚需进一步研究[288],异黄酮上调miR-200和let-7导致吉西他滨耐药PDAC细胞EMT逆转,这可能对设计吉西他滨为基础的PDAC新疗法很重要[289]。重要的是,在人类PDAC的正交各向异性模型中,Harikumaret al。[290]发现白藜芦醇显著抑制肿瘤生长,吉西他滨进一步增强了这种作用。作者还证实,白藜芦醇可以通过抑制增殖、侵袭、血管生成和转移的标记物来增强吉西他滨的作用[290]。Arshad报道等.[291]研究表明,静脉注射100g海洋源ω-脂肪酸(ω-FA)联合吉西他滨可改善晚期胰腺癌患者的活性和生活质量。这项吉西他滨和ω-FA输注的研究数据表明,47.2%的患者在全球健康状况上有10%或以上的改善,这被认为是具有临床意义和令人鼓舞的[291]。ESPAC-3试验,III期随机对照试验,在159例胰腺癌中进行,以确定5-FU或吉西他滨作为胰腺癌切除术后辅助治疗在OS方面是否优于前者(clinicaltrials.gov标识:NCT00058201)[292]。患者服用叶酸(20mg/m)2)与5-FU或吉西他滨联合使用[292]。治疗组间PFS和整体生活质量评分均无显著差异[292]。与使用氟尿嘧啶加叶酸相比,吉西他滨对完全切除的胰腺癌患者的OS没有改善[292]。
最近,程et al。[293]表明美异靛蓝对吉西他滨耐药PDACs具有抗增殖作用,其化学性质与天然产物靛玉红有关。特别是,研究发现具有干细胞表型的细胞更容易受到美isoindigo的影响,美isoindigo会减少csc相关基因的表达,降低细胞的流动性和球的形成,降低葡萄糖的吸收,同时增加ROS水平[293]。
山奈酚(银杏黄酮)抑制PDAC细胞增殖并诱导凋亡,并可能使细胞对5-FU敏感,因为它们的联合给药显示出抑制增殖的加性效应[294]。值得注意的是,山奈酚在正常人胰腺导管上皮细胞中的细胞毒性明显低于5-FU[294]。此外,当山奈酚以浓度依赖性的方式处理时,凋亡细胞数量增加[294]。
最近的研究表明,十字花科蔬菜中的一组关键活性成分异硫氰酸酯可能是PARP治疗的补充治疗机制[295]。Banerjee的体外研究等.[296]研究表明,用大豆源染料木素预处理细胞后再用顺铂预处理细胞会导致细胞活力的显著丧失和细胞凋亡的增强,而与细胞的转移能力无关。此外,作者在其原位肿瘤模型中发现,染料木素联合顺铂比单独使用顺铂更有效的抗肿瘤药物[296]。染料木素通过抑制Bcl-xL、Bcl-2和AKT激酶和激活NF-κB使胰腺癌细胞对顺铂诱导的凋亡敏感[294,296]。
VDR的激活是细胞毒性药物敏感性的关键决定因素,因为它对修复停滞的复制叉非常重要[297]。增敏机制是通过募集同源重组中的关键蛋白RAD51[297]。虽然VDR的敲除增强了吉西他滨的杀伤作用,但PDAC细胞中VDR表达和激活水平的增加增加了其对吉西他滨的IC50[297]。VDR对吉西他滨敏感性的影响依赖于配体和二聚体,缺乏这些活性的VDR突变体对吉西他滨敏感性无效[297]。抑制PDAC中的VDR为提高PARP抑制剂和吉西他滨等基因毒性药物的疗效提供了一种途径。相比之下,Persons等[298]表明,在胰腺癌细胞中,维生素D的烷基化衍生物(1,25(OH)2D3),维生素D的抗增殖特性在与AMPK通路激活剂AICAR共同给药时得到了强烈增强。因此,在癌症治疗中使用特定的天然或合成化合物需要深入了解癌症信号通路和网络之间的相互作用,以保留或增强化学预防活性或增强化疗试剂的有效性,同时减少已知的毒性作用。
时间疗法依赖于癌症治疗计划的充分昼夜节律,通过增强抗癌药物的耐受性和疗效,在实验和临床研究中都提供了令人鼓舞的结果。将时间生物学概念应用于PDAC的治疗似乎是有效的,因为与用于PDAC术后辅助治疗的基于时间调节的5-FU输注放化疗相比,其急性毒性的严重程度和频率相对较低[299]。然而,最佳时机因性别、基因背景、生活方式、癌症分期和是否患有其他疾病而异。在这种程度上,与药物代谢、细胞解毒、细胞周期和昼夜节律节律特征相关的生物标志物可能有助于优化给药时机。分别报道了昼夜节律基因在PDAC中的诊断和预后潜力,其临床应用有待在更大人群中验证(表6)[300]。Oda的支持性报告等[301]。建议的昼夜节律基因PER2因为昼夜节律基因的过表达PER2.PER2基因编码周期昼夜节律蛋白同源物2,该蛋白作为肿瘤抑制基因,与顺铂具有协同作用(表6)。正常昼夜节律周期的破坏,导致昼夜节律基因的失调,已与多种疾病相关,如肥胖、糖尿病、炎症、睡眠障碍癌症,包括PDAC[300,301]。因此,重新编程生物钟可能为开发新的策略提供很大的机会,旨在治疗或预防癌症过程中的生物钟功能障碍。值得注意的是,在哺乳动物PDAC模型中,进餐时间被证明可以诱导关键基因的节律性表达,并抑制肿瘤的生长,尽管分子时钟没有功能[302]。似乎将食物限制在黑暗期、合理的睡眠周期、有计划的锻炼、健康的饮食(例如,多吃水果、蔬菜和全谷物)可能有效地挽救生物钟,帮助纠正代谢功能障碍。这种新颖的基于昼夜节律的支持性护理值得临床试验。
总之,PDAC继续构成一个主要的治疗挑战,因为即使在那些有良好表现状态并适合更强化治疗方案的患者中,预期寿命也很少超过12个月。这种失败在很大程度上是由于PDAC的病理特性,可以合理地推测,更好地了解PDAC的生物学可能导致制定有效的管理计划。重要的是,更好地了解肿瘤生物学以及PDAC的遗传易感性、基因-基因相互作用、基因-环境相互作用和表观遗传现象,将有助于识别新的靶点和相关途径,作为诊断、预测和预后的生物标志物。多亏了新技术和基于“组学”的特征研究,分子分类PDACs正在迅速发展,不仅能识别多个新靶点,还能识别可能对特定治疗组合有反应的患者子集。目前,手术提供了最好的生存机会,因此,发现准确的生物标记物,以早期诊断和识别最有可能从手术和新辅助治疗获益的患者亚群,可能会增加可切除患者的数量。由于PDAC在诊断时已经是一种系统性疾病,PDAC的管理需要一个移动和动态的规划,可以从患者肿瘤的多区域采样和药物组合开始,每种药物针对不同的亚克隆和微环境室的特征,然后使用循环生物标志物和成像方法实时监测疾病。考虑到目前常规化疗的局限性,包括癌症患者的严重毒性和生活质量下降,开发安全有效的补充或替代干预措施,如已知/可预测的作用机制的天然产物,似乎可以提高治疗效果,同时改善生活质量。进一步的临床前研究和精心设计的临床试验是非常必要的,以加速开发为每个患者提供最佳治疗的新策略。基于生物标志物的生活方式建议的个性化医疗方法可能提供一种新的方法来评估PDAC患者的健康,向他们提供他们需要的信息来重新控制他们的生活。希望这些方法的结合将在某种程度上改善患有这种毁灭性疾病的个体的生活质量和生存。
这篇文章是为了纪念乔恩·洛德。我们感谢葵花果酱慈善机构将Jon Lord奖学金授予PK。我们也要感谢Meleni Aldridge (ANH国际)在整个项目期间的鼓励和支持。
- 2012年全球癌症发病率和死亡率:国际癌症研究机构癌症基地第11号法国里昂:国际癌症研究机构。
- Klöppel G(2000)世界卫生组织《消化系统肿瘤的肿瘤分类、病理学和遗传学:胰腺导管腺癌》,国际癌症研究机构出版社。
- 英国癌症研究(2014)胰腺癌死亡率统计。
- Siegel RL, Miller KD, Jemal A(2016)癌症统计2016。CA癌症J临床66: 7-30。Crossref]
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM,等(2014)预测2030年癌症发病率和死亡率。在美国,甲状腺癌、肝癌和胰腺癌的意外负担。癌症Res74: 2913 - 2921。[Crossref]
- Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N,等(2016)当前晚期胰腺癌的治疗策略:临床医师综述。世界J clinical Oncol7: 27-43。[Crossref]
- 胡特曼,de Ondarza J(1993)胰管生理学和病理生理学综述。消化54: 323 - 330。(Crossref)
- Otsuki M, Tashiro M(2007)慢性胰腺炎和胰腺癌,生活方式相关疾病。地中海的实习生46: 109 - 113。[Crossref]
- Howlader N (2015) SEER癌症统计评论,1975-2012,国家癌症研究所。
- Maisonneuve P, Lowenfels AB(2015)胰腺癌的危险因素:荟萃分析研究综述。国际流行病学44: 186 - 198。(Crossref)
- Becker AE, Hernandez YG, Frucht H, Lucas AL(2014)胰腺导管腺癌:危险因素、筛查和早期发现。世界J肠胃醇20: 11182 - 11198。[Crossref]
- Bartsch DK, Gress TM, Langer P(2012)家族性胰腺癌现状知识。Nat Rev胃肠醇9: 445 - 453。[Crossref]
- Aichler M, Seiler C, Tost M, Siveke J, Mazur PK,等。(2012)转基因小鼠与人组织中非典型扁平病变的胰腺导管腺癌起源的比较研究。中草药226年,723 - 734。[Crossref]
- Hruban RH, Klimstra DS(2014)胰腺腺癌。Semin诊断病理科31日:443 - 451。(Crossref)
- Erkan M, Michalski CW, Rieder S, reier -Erkan C, Abiatari I,等(2008)激活间质指数是一种新的独立的胰腺导管腺癌预后标志物。临床胃肠醇6: 1155 - 1161。[Crossref]
- Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M,等(2012)间质在胰腺癌中的作用:诊断和治疗意义。Nat Rev胃肠醇9: 454 - 67。[Crossref]
- Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE等(2009)546例胰腺神经病变和神经痛的综合病理研究。胃肠病学139: 177 - 186。[Crossref]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ,等(2008)全球基因组分析揭示的人类胰腺癌核心信号通路。科学32: 1801 - 1806。[Crossref]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, et al.(2015)全基因组重新定义胰腺癌的突变格局。自然518: 495 - 501。[Crossref]
- 王晓燕,王晓燕,王晓燕,等(2008)家族性胰腺癌的遗传和表观遗传改变。癌症流行病学生物标志物17: 3536 - 3542。[Crossref]
- Kanda M, matthaai H, Wu J, Hong SM, Yu J,等(2012)大多数早期胰腺上皮内瘤变存在体细胞突变。胃肠病学142: 730 - 733。[Crossref]
- Eser S, Schnieke A, Schneider G, Saur D(2014)胰腺癌的致癌KRAS信号通路。乳腺癌111: 817 - 822。(Crossref)
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S,等(2012)小鼠胰腺癌的起始和维持都需要致癌Kras。J clinin投资122: 639 - 653。(Crossref)
- Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J,等(2012)小鼠转移性胰腺癌依赖于致癌Kras。《公共科学图书馆•综合》7: e49707。(Crossref)
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB,等(2005)Trp53R172H和KrasG12D协同促进小鼠胰腺导管腺癌的染色体不稳定性和广泛转移。癌症细胞7: 469 - 483。[Crossref]
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE,等(2012)RAF - >MEK - >ERK信号通路在胰腺导管腺癌发生中的中心作用。癌症越是加大2: 685 - 693。[Crossref]
- Grover S, Jajoo K(2016)高危人群胰腺癌筛查。美国北部胃肠科诊所45: 117 - 127。(Crossref)
- Reznik R, Hendifar AE, Tuli R(2014)胰腺腺癌的遗传决定因素和潜在的治疗靶点。前面的杂志5: 87。[Crossref]
- Morris JP, Wang SC, Hebrok M (2010) KRAS, Hedgehog, Wnt与胰腺导管腺癌的扭曲发育生物学。Nat Rev癌症10: 683 - 695。[Crossref]
- Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D,等(2013)高脂饮食激活致癌性Kras和COX2诱导小鼠胰腺导管腺癌的发展。胃肠病学145: 1449 - 1458。[Crossref]
- White, E(2013)利用rasdriven cancer的不良饮食习惯。基因开发27日:2065 - 2071。[Crossref]
- Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M(2013)炎症、自噬和肥胖:胰腺炎和胰腺癌发病的共同特征。胃肠病学144年,1199 - 1209。[Crossref]
- Karna E, Surazynski A, orowski K, Łaszkiewicz J, Puchalski Z,等(2002)血清和组织中胰岛素样生长因子- i (IGF-I)和IGF-I结合蛋白作为胰腺炎和胰腺癌的指标。Int J Exp Pathol83: 239 - 245。[Crossref]
- Appleman VA, Ahronian LG, Cai J, Klimstra DS, Lewis BC (2012) KRAS(G12D)和BRAF(V600E)诱导的小鼠胰腺上皮细胞转化需要MEK/ erk刺激的IGF1R信号。Mol Cancer Res10: 1228 - 1239。[Crossref]
- Tanno S, Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH,等(2001)AKT激活上调胰岛素样生长因子I受体表达并促进人胰腺癌细胞的侵袭性。癌症Res61年589 - 593。[Crossref]
- Ruggeri BA,黄亮,Wood M,程建强,Testa JR(1998)人胰导管腺癌中AKT2癌基因的扩增和过表达。摩尔Carcinog2:81 - 86。[Crossref]
- Asano T,姚颖,朱杰,李迪,Abbruzzese JL,等(2005)胰腺癌细胞中Pten异常表达激活pi3 -激酶/Akt信号通路,靶向转录因子NF- kappa B和c-Myc。致癌基因23日:8571 - 8580。[Crossref]
- Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G,等(2014)靶向mTOR在胰腺癌中的依赖性。肠道63: 1481 - 1489。(Crossref)
- 冯铮(2010)p53对IGF-1/AKT/mTOR通路和内体间室的调控。冷泉港远景生物2: a001057。(Crossref)
- Rozengurt E, Soares HP, Sinnet-Smith J(2014)抑制由PI3K/mTOR介导的反馈循环可诱导代偿通路的多重过度激活:导致耐药性的意外后果。Mol Cancer Ther13: 2477 - 2488。[Crossref]
- Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, et al.(2002)胰岛素/胰岛素样生长因子I杂交受体具有不同的生物学特性,这取决于所涉及的胰岛素受体亚型。J生物化学277: 39684 - 39695。[Crossref]
- Rozengurt E, sinnet - smith J, Kisfalvi K(2010)胰岛素/胰岛素样生长因子-1受体和G蛋白偶联受体信号系统之间的串扰:抗糖尿病药物amm在胰腺癌中的一个新靶点。临床癌症治疗16: 2505 - 2511。[Crossref]
- 上田雪梅,王晓燕,王晓燕,等。胰岛素样生长因子受体1型与表皮生长因子受体在胰腺癌进展和转移中的相互作用。国防部分册19日:788 - 796。[Crossref]
- Ucar DA, Magis AT, He DH, Lawrence NJ, Sebti SM等(2013)抑制cMET和IGF-1R与FAK的相互作用可有效降低胰腺癌细胞的体外和体内生长。抗癌药物医药化学13: 595 - 602。[Crossref]
- Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R(2009)胰岛素受体异构体和胰岛素受体/胰岛素样生长因子受体在生理和疾病中的杂交。Endocr牧师30: 586 - 623。[Crossref]
- Ulanet DB, Ludwig DL, Kahn CR, Hanahan D.(2010)胰岛素受体在功能上促进了肿瘤的多阶段进展,并传递了对IGF-1R靶向治疗的内在抵抗。PNAS107: 10791 - 10798。[Crossref]
- Morton JP, Jamieson NB, Karim SA, Athineos D, Ridgway RA, et al. (2010) LKB1单倍不足与Kras合作,通过抑制p21依赖的生长阻滞促进胰腺癌。胃肠病学139: 586- 97,597 .e1-6。[Crossref]
- Cheng X, Kim JY, Ghafoory S, Duvaci T, Rafiee R, et al.(2014)甲基异蓝靛通过抑制pdac中LKB1和激活AMPK干扰细胞代谢,优先杀死癌症干细胞。摩尔杂志[Crossref]
- Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E(2009)二甲双胍干扰G蛋白偶联受体和胰岛素受体信号系统之间的串扰,抑制胰腺癌生长。癌症Res69: 6539 - 6545。[Crossref]
- Oliveira-Cunha M, Newman WG, Siriwardena AK(2011)胰腺癌中的表皮生长因子受体,癌症3:1513 - 1526。[Crossref]
- Sánchez-González P, Jellali K, Villalobo A(2010)钙调素介导的表皮生长因子受体调控。2月J277: 327 - 342。[Crossref]
- Tobita K, Kijima H, Dowaki S, Kashiwagi H, Ohtani Y,等。(2003)表皮生长因子受体在人胰腺癌中的表达:肝转移的意义。Int J Mol医学1: 305 - 309。[Crossref]
- 高金,谢克华,李志刚,等(1998)胰胆管腺癌转化生长因子β受体基因的遗传改变。癌症Res58: 5329 - 5332。[Crossref]
- Schutte M, hurban RH, Geradts J, Maynard R, Hilgers W,等(1997)Rb/p16肿瘤抑制通路在几乎所有胰腺癌中的撤销。癌症Res57: 3126 - 3130。[Crossref]
- Kim WY, Sharpless NE (2006) INK4/ARF在癌症和衰老中的调节作用。细胞127: 265 - 275。(Crossref)
- Ruggeri BA, Huang L, Berger D, Chang H, Klein-Szanto AJ,等(1997)原发性和转移性胰腺导管病变的分子病理学:散发性和家族性病变中p53、mdm-2和p21/ waf1基因的突变和表达分析。癌症79: 700 - 716。[Crossref]
- Carnevale J, Ashworth A2(2015)评估胰腺癌中BRCA1和BRCA2突变的意义。J clinin Oncol33: 3080 - 3081。(Crossref)
- Powell SN, Kachnic LA (2003) BRCA1和BRCA2在同源重组、DNA复制保真度和细胞对电离辐射的响应中的作用。致癌基因22日:5784 - 5791。[Crossref]
- Sy SM, Huen MS, Chen J (2009) PALB2是BRCA复合体中不可或缺的组成部分,需要进行同源重组修复。美国国家科学研究院106: 7155 - 7160。[Crossref]
- mcabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A,等(2006)同源重组修复DNA损伤的缺陷和对聚(adp -核糖)聚合酶抑制的敏感性。癌症Res66: 8109 - 8115。[Crossref]
- 董霞,李颖,Hess KR, Abbruzzese JL,李迪(2011)DNA错配修复基因多态性对胰腺癌患者生存的影响。肿瘤学家16: 61 - 70。(Crossref)
- swetach P, Hulikova A, Vaughan-Jones RD, Harris AL.(2000)对碳酸酐酶IX在肿瘤pH调节中的生理作用的新认识。致癌基因29日:6509 - 6521。[Crossref]
- Dovmark TH, Hulikova A, Vaughan-Jones RD, Swietach P(2014)三维胰腺癌生长过程中碳酸酐酶活性调节细胞内pH值。Proc physics Soc3: PCA114(摘要)。
- Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH,等(2013)肿瘤微环境产生的酸性驱动局部浸润。癌症Res73: 524 - 1535。[Crossref]
- Kong SC, Giannuzzo A, Novak I, Pedersen SF(2014)胰腺癌中酸碱转运的分子机制和临床潜力.生物化学细胞生物学92: 449 - 459。[Crossref]
- Cardone RA, Greco MR, Zeeberg K1, Zaccagnino A2, Saccomano M等(2015)一种新型的以nhe1为中心的信号盒子驱动表皮生长因子受体依赖性的胰腺肿瘤转移,是联合治疗的靶点。瘤形成17: 155 - 166。[Crossref]
- Yee NS, Chan AS, Yee JD, Yee RK(2012)胰腺腺癌中的TRPM7和TRPM8离子通道:作为癌症生物标志物和靶点的潜在作用。Scientifica(开罗)2012: 415158。[Crossref]
- 董宏,沈金坤,李建明,Estrema C, Ornelas TA,等(2010)Ca2+介导的人胰管细胞运动的分子机制。Am J Physiol Cell Physiol299: 1493 - 1503。[Crossref]
- Ahmed N, Corey M, Forstner G, Zielenski J, Tsui LC,等。(2003)外分泌胰腺中囊性纤维化跨膜传导调节因子(CFTR)基因突变的分子后果。肠道52: 1159 - 1164。[Crossref]
- Neureiter D, Jäger T, Ocker M, Kiesslich T(2014)表观遗传学与胰腺癌:病理生理学和新的治疗方面。世界J肠胃醇20: 7830 - 7848。(Crossref)
- Visani M, Acquaviva G, Fiorino S, Bacchi Reggiani ML, Masetti M,等(2015)microRNA分析对胰腺病变特征的贡献:综述。临床病理学杂志68: 859 - 869。(Crossref)
- Fukushige S,堀井A(2014)胰腺癌早期检测之路:尝试利用表观遗传生物标志物。癌症列托人342: 231 - 237。[Crossref]
- Reya T, Morrison SJ, Clarke MF, Weissman IL(2001)干细胞,癌症,和癌症干细胞。自然414: 105 - 111。(Crossref)
- 李超,李cj, Simeone DM(2009)人胰腺癌干细胞的鉴定。方法Mol Biol568: 161 - 173。(Crossref)
- Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, et al. (2012) Hedgehog-EGFR协同反应基因决定了基底细胞癌和肿瘤启动性胰腺癌细胞的致癌表型。摩尔医学4: 218 - 33所示。[Crossref]
- Jimeno A, Feldmann G, Suárez-Gauthier A, Rasheed Z, Solomon A, et al.(2009)一种直接胰腺癌异种移植模型作为癌症干细胞治疗开发的平台。Mol Cancer Ther8: 310 - 314。[Crossref]
- 李超,吴俊杰,Hynes M, Dosch J, Sarkar B,等(2011)C - met是胰腺癌干细胞的标记物和治疗靶点。胃肠病学141: 2218 - 2227。(Crossref)
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW,等(2007)不同的癌症干细胞群决定了人类胰腺癌的肿瘤生长和转移活性.细胞干细胞313 - 323。[Crossref]
- BA老师,Fricker SP(2010)癌症中的CXCL12 (SDF-1)/CXCR4通路。临床癌症治疗16: 2927 - 2931。(Crossref)
- Salnikov AV, Liu L, Platen M, Gladkich J, Salnikova O, et al.(2012)低氧诱导低侵袭性和高侵袭性胰腺肿瘤细胞EMT,但只有具有肿瘤干细胞特征的细胞具有明显的迁移潜能。《公共科学图书馆•综合》7: e46391。[Crossref]
- Rausch V, Liu L, Apel A, Rettig T, Gladkich J,等。(2012)自噬介导低氧微环境下胰腺肿瘤启动细胞的存活。中草药227:325 - 335(2012)。[Crossref]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N,等(2014)癌基因耐蚀性胰腺癌细胞依赖于线粒体功能。自然514: 628-632 [Crossref]
- Inman KS, Francis AA, Murray NR(2014)免疫系统在胰腺癌发生和发展中的复杂作用。世界J肠胃醇20: 11160 - 11181。[Crossref]
- Clark CE, Beatty GL, Vonderheide RH(2009)胰腺腺癌的免疫监测:来自基因工程小鼠癌症模型的见解。癌症列托人279: 1 - 7。[Crossref]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM,等(2012)EMT和播散先于胰腺肿瘤的形成。细胞148: 349 - 361。(Crossref)
- 谢东,谢k1(2015)胰腺癌基质生物学与治疗。基因说2: 133 - 143。(Crossref)
- 亚av D, Lowenfels AB(2013)胰腺炎与胰腺癌的流行病学研究。胃肠病学144: 1252 - 1261。(Crossref)
- Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M(2013)炎症、自噬和肥胖:胰腺炎和胰腺癌发病的共同特征。胃肠病学144: 1199 - 1209。[Crossref]
- Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI,等(2016)布鲁顿酪氨酸激酶(BTK)依赖性免疫细胞串声驱动胰腺癌。癌症越是加大6: 270 - 285。[Crossref]
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, et al. (2011) STAT3在kras诱导的胰腺肿瘤发生中发挥关键作用。癌症Res71: 5020 - 5029。(Crossref)
- 埃文斯,科斯特洛E(2012)炎症细胞在促进胰腺癌细胞生长和侵袭中的作用。前面的杂志3: 270。(Crossref)
- addemmer K, Ebert M, Müller-Ostermeyer F, Friess H, Büchler MW等(1998)人胰腺癌中的效应T淋巴细胞亚群:通过多表位显像检测CD8+CD18+细胞和CD8+CD103+细胞。临床试验免疫112:。第21到26[Crossref]
- Hiraoka N, Onozato K, Kosuge T, Hirohashi S (2006) FOXP3+调节性T细胞在胰腺导管腺癌及其癌前病变进展过程中增加.临床癌症治疗12: 5423 - 5434。[Crossref]
- Husain Z, Huang Y, Seth P, Sukhatme VP(2013)肿瘤源性乳酸修饰抗肿瘤免疫反应:对骨髓源性抑制细胞和NK细胞的影响。J Immunol19:1486 - 1495。[Crossref]
- Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M(2013)炎症、自噬和肥胖:胰腺炎和胰腺癌发病的共同特征。胃肠病学144: 1199 - 1209。[Crossref]
- 杨志强,李志强,李志强,等(2005)胰腺癌患者外周血树突状细胞功能的变化。中国Immunol114: 52-60。[Crossref]
- Ryschich E, Nötzel T, Hinz U, Autschbach F, Ferguson J,等(2005)人胰腺癌HLA - I类对T细胞介导的免疫应答的控制。临床癌症治疗1: 498 - 504。[Crossref]
- von Bernstorff W, Spanjaard RA, Chan AK, Lockhart DC, Sadanaga N,等(1999)胰腺癌细胞可以通过非功能性Fas (APO-1/CD95)受体和功能性Fas配体的异常表达逃避免疫监视。Syrgery125: 73 - 84。[Crossref]
- Suzuki N, Maeda Y, Tanaka S, Hida N, Mine T,等(2002)用于胰腺癌特异性免疫治疗的肽特异性细胞毒性T淋巴细胞前体的检测。Int J癌症98: 45 - 50。
- Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y等(2004)CD8+肿瘤浸润淋巴细胞联合CD4+肿瘤浸润淋巴细胞和树突状细胞改善胰腺腺癌患者的预后。胰腺28: 26-31。[Crossref]
- Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, et al.(1998)大鼠胰腺腺泡周星状细胞的鉴定、分离和培养。肠道43: 128 - 133。[Crossref]
- 周松,周晓燕,周晓燕,周晓燕(2008)胰腺星状细胞在胰腺癌中的作用。兰根贝克拱门外科医生393: 891 - 900。(Crossref)
- Apte MV, Park S, Phillips PA, Santucci N, Goldstein D,等(2004)胰腺星状细胞在胰腺癌中的粘连增生反应。胰腺29:179e87。[Crossref]
- Apte MV, Wilson JS, Lugea A, Pandol SJ(2013)星形细胞在胰腺癌微环境中的主要作用。胃肠病学144: 1210 - 1219。[Crossref]
- Apte MV, Xu Z, Pothula S, Goldstein D, Pirola RC,等(2015)胰腺癌:微环境也需要关注!Pancreatology15: S32-38。(Crossref)
- 福岛N,菊池Y, Nishiyama T, Kudo A, Fukayama M(2008)胰腺侵袭性和导管内肿瘤间质中的骨膜沉积。国防部分册2: 1044 - 1053。[Crossref]
- Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, et al.(2013)胰腺癌相关星状细胞以stat3依赖的方式促进骨髓源性抑制细胞的分化。癌症Res73: 3007 - 3018。[Crossref]
- Mace TA, Bloomston M, Lesinski GB(2013)胰腺癌相关星状细胞:肿瘤微环境中减少免疫抑制的可行靶点。Oncoimmunology2: e24891。[Crossref]
- Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R,等(2013)激活的胰腺星状细胞隔离CD8+ t细胞以减少其对胰腺导管腺癌瘤旁室的浸润。胃肠病学145: 1121 - 1132。[Crossref]
- 高智,王旭,吴坤,赵莹,胡刚(2010)胰腺星状细胞通过基质细胞衍生因子-1/CXCR4轴增加人胰腺癌细胞的侵袭。Pancreatology10: 186 - 193。[Crossref]
- 徐铮(2010)胰腺星状细胞在胰腺导管腺癌转移中的作用。Am J Pathol177:2585 - 2596。
- 田永伟,吴艳梅,林伟武,李海生,乐海生(2009)胰腺癌细胞刺激肝星状细胞增殖和基质合成。J乙醇5: 307 - 314。
版权所有OAT。版权所有- Kordes C, Sawitza I, Häussinger D(2009)肝脏和胰腺星状细胞的焦点。临床生物化学390: 1003 - 1012。(Crossref)
- 李霞,王铮,马强,徐强,刘宏等(2014)超音hedgehog基因旁分泌信号激活间质细胞促进胰腺癌神经周侵袭。临床癌症治疗20: 4326 - 4338。[Crossref]
- Preston M, Sherman LS(2011)神经干细胞龛:透明质酸基细胞外基质的作用。前面Biosci3:1165 - 1179。[Crossref]
- Schuller HM(2008)神经传递和癌症:预防和治疗的意义。抗癌药物19日:655 - 671。(Crossref)
- 朱铮,Kleeff J, Kayed H, Wang L, Korc M,等(2002)神经生长因子对胰腺癌细胞增殖、侵袭和致瘤性的促进作用。摩尔Carcinog35: 138 - 147。[Crossref]
- Lunardi S, Muschel RJ, Brunner TB(2014)胰腺癌的间质间室:有任何治疗靶点吗?癌症列托人343: 147 - 55。
- Ottaviano AJ, Sun L, Ananthanarayanan V, Munshi HG(2006)胰腺导管细胞中细胞外基质介导的膜型1基质金属蛋白酶表达受转化生长因子-ß1调控。癌症Res66: 7032 - 7040。[Crossref]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD(2012)间质酶靶向消除胰腺导管腺癌治疗的物理障碍。癌症细胞2: 418 - 429。[Crossref]
- 徐勇,Baba H, Fukuda T, Takashima M, Sugimachi K(2000)导管胰腺腺癌患者血管内皮生长因子高表达与肝转移和不良预后相关。癌症8: 2239 - 2245。[Crossref]
- 王海霞,陈庆坤(2003)[环氧合酶-2在人胰腺癌组织中的表达及意义]。Ai郑22日:649 - 652。(Crossref)
- Fukahi K, Fukasawa M, Neufeld G, Itakura J, Korc M(2004)人胰腺癌细胞中神经素-1和-2的异常表达。临床癌症治疗10: 581 - 590。(Crossref)
- 在胰腺导管腺癌中,癌症-星状细胞相互作用延续了缺氧-纤维化周期。瘤形成1:497 - 508。
- Mclntyre CA, Winter JM(2015)胰腺导管腺癌的诊断评价和分期。Semin杂志42: 19-27。[Crossref]
- 李丹(2012)糖尿病与胰腺癌。摩尔Carcinog51: 64 - 74。(Crossref)
- Mayr M, Schmid RM(2010)胰腺癌和抑郁症:神话和真相。BMC癌症10: 569。(Crossref)
- Apple F, Benson P, Preese L, Eastep S, Bilodeau L,等(1991)组织和高淀粉酶血症患者的脂肪酶和胰淀粉酶活性。J是Clin Pathol吗96: 610 - 614。(Crossref)
- Winter JM, Yeo CJ, Brody JR(2013)胰腺癌的诊断、预后和预测性生物标志物。外科医师107:第15 - 22。(Crossref)
- Al-Hawary MM, Francis IR(2013)胰腺导管腺癌分期。癌症成像13: 360 - 364。(Crossref)
- Edge SB, Compton CC(2010)美国癌症联合委员会:AJCC癌症分期手册第7版和TNM的未来。安外科医生17: 1471 - 1474。[Crossref]
- Tempero MA, Arnoletti JP, Behrman SW, Ben-Josef E, Benson AB 3rd,等人(2012)胰腺腺癌,版本2.2012:NCCN指南的特征更新。[中文]10: 703 - 713。(Crossref)
- Vauthey JN, Dixon E (2009) AHPBA/SSO/SSAT可切除和边缘可切除胰腺癌共识会议:会议原理和概述。安外科医生16: 1725 - 1726。[Crossref]
- 转移性胰腺癌的病理学和遗传学研究。Arch病理科实验室133: 413 - 422。(Crossref)
- Hidalgo M, Cascinu S, Kleeff J, Labianca R, Löhr JM,等(2015)解决胰腺癌的挑战:改善预后的未来方向。Pancreatology15:仅。[Crossref]
- Sclafani F, Iyer R, Cunningham D, Starling(2015)转移性胰腺癌的管理:当前的治疗方案和潜在的新的治疗靶点。暴击恢复血液病95: 318 - 336。[Crossref]
- Neoptolemos JP, Dunn JA, Stocken DD, Almond J, Link K,等。(2001)可切除胰腺癌的辅助放化疗:一项随机对照试验。《柳叶刀》358: 1576 - 1585。[Crossref]
- [未列出作者](1987)GITSG。胰腺癌根治切除后有效辅助放疗和化疗的进一步证据。胃肠道肿瘤研究组。癌症59: 2006 - 2010。[Crossref]
- Klinkenbijl JH, Jeekel J, sahoud T, van Pel R, Couvreur ML,等(1999)胰腺和壶腹周围癌治愈切除后辅助放疗和5-氟尿嘧啶:EORTC胃肠道肿瘤合作组的III期试验。安杂志230: 776 - 782。[Crossref]
- Neoptolemos JP, Stocken DD, Dunn JA, Almond J, Beger HG,等(2001)ESPAC-1随机对照试验中辅助放化疗和/或化疗胰腺癌患者切缘对生存期的影响。安杂志234: 758 - 768。[Crossref]
- Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA,等(2004)胰腺癌切除术后放化疗的随机试验。N英语J医学350: 1200 - 1210。(Crossref)
- Oettle H(2013)吉西他滨辅助化疗与胰腺癌切除患者的长期预后:CONKO-001随机试验。《美国医学会杂志》310: 1473 - 1481。[Crossref]
- Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D,等(2010)胰腺癌切除术后氟脲嘧啶+叶酸辅助化疗vs吉西他滨:一项随机对照试验。《美国医学会杂志》304: 1073 - 1081。[Crossref]
- Regine W(2006)一项III期组间试验(RTOG 97-04):辅助化疗前后(CRT) 5-FU与吉西他滨(G)对切除胰腺腺癌的疗效比较。放射肿瘤学医学杂志理论物理66: 23 - 24日。
- Katz MH1, Pisters PW, Evans DB, Sun CC, Lee JE,等(2008)边缘可切除胰腺癌:这一疾病新发阶段的重要性。J Am Coll外科医生206: 833 - 848。[Crossref]
- Abrams RA1, Lowy AM, O'Reilly EM, Wolff RA, Picozzi VJ等(2009)可切除和边缘可切除胰腺癌的联合治疗:专家共识声明。安外科医生16: 1751 - 1756。[Crossref]
- Sclafani F, Iyer R, Cunningham D, Starling N(2015)转移性胰腺癌的管理:当前的治疗方案和潜在的新的治疗靶点。暴击恢复血液病95: 318 - 336。[Crossref]
- Kim HW, Lee JC, Paik KH, Lee YS, Hwang JH, et al.(2015)最初转移部位作为IV期胰导管腺癌患者的预后因素。医学(巴尔的摩)94: e1012。[Crossref]
- Huguet F, Mukherjee S2, Javle M3(2014)局部晚期胰腺癌:确定性放化疗的作用。Clin Oncol (R Coll Radiol)26日:560 - 568。(Crossref)
- Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML,等(1997)吉西他滨作为晚期胰腺癌患者一线治疗的生存改善和临床获益:一项随机试验。J clinin Oncol15: 2406 - 2413。[Crossref]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR等(2007)厄洛替尼加吉西他滨与单独吉西他滨在晚期胰腺癌患者中的疗效比较:加拿大国家癌症研究所临床试验组的一项III期试验。J clinin Oncol25:1960 - 1966。[Crossref]
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R,等(2011)FOLFIRINOX与吉西他滨治疗转移性胰腺癌。N英语J医学364: 1817 - 1825。(Crossref)
- Ho MY, Kennecke HF, Renouf DJ,张WY, Lim HJ等(2015)在不列颠哥伦比亚省确定FOLFIRINOX对一线转移性胰腺腺癌(MPC)的适用性:一项基于人群的回顾性研究.J是clinin Oncol吗[Crossref]
- Mahaseth H, Brutcher E, Kauh J, Hawk N, Kim S(2013)改进FOLFIRINOX方案,提高了胰腺癌的安全性并保持了疗效。胰腺42: 1311 - 1315。[Crossref]
- Gunturu KS, Yao X, Cong X, Thumar JR, Hochster HS等(2013)FOLFIRINOX治疗局部晚期和转移性胰腺癌:单机构回顾性研究疗效和毒性。医学杂志30: 361。[Crossref]
- Blazer M, Wu C, Goldberg RM, Phillips G, Schmidt C,等。(2015)新辅助剂改良FOLFIRINOX治疗局部晚期不可切除(LAPC)和边缘可切除(BRPC)胰腺腺癌。安外科医生22日:1153 - 1159。[Crossref]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J等(2013)nabu -紫杉醇联合吉西他滨可提高胰腺癌患者的生存率。N英语J医学369: 1691 - 1703。(Crossref)
- Al-Hajeili M, Azmi AS2, Choi M3 (2014) nab -紫杉醇治疗晚期胰腺癌的潜力。Onco目标Ther7: 187 - 192。(Crossref)
- Pelzer U, Schwaner I, Stieler J, Adler M, Seraphin J等(2011)在二线晚期胰腺癌患者中最佳支持护理(BSC)与奥沙利铂、叶酸和5-氟尿嘧啶(OFF)加BSC的比较:来自德国conko研究组的一项III期研究。Eur J癌47: 1676 - 1681。[Crossref]
- Berger AK, Weber TF, Jäger D, Springfeld C(2013)一例转移性胰腺腺癌患者FOLFIRINOX失败后成功应用nab-紫杉醇和吉西他滨治疗。肿瘤学杂志,36,763-765。[Crossref]
- Portal A, Pernot S, Tougeron D, Arbaud C, Bidault AT等人(2015)nag -紫杉醇联合吉西他滨治疗Folfirinox失败后的转移性胰腺腺癌:AGEO前瞻性多中心队列研究。乳腺癌113年,989 - 995。[Crossref]
- Zhang Y, Hochster H, Stein S, Lacy J(2015)吉西他滨联合nab-紫杉醇治疗一线FOLFIRINOX晚期胰腺癌:单机构回顾性研究疗效和毒性。Exp血液病29。Crossref]
- da Rocha Lino A, Abrahão CM, Brandão RM, Gomes JR, Ferrian AM,等(2015)晚期胰腺癌FOLFIRINOX进展后吉西他滨作为二线治疗的作用:回顾性分析。胃肠病学杂志6: 511 - 51。[Crossref]
- Wang-Gillam A, Li CP2, Bodoky G, Dean A, Shan YS,等(2016)纳米脂质体伊立替康联合氟尿嘧啶和叶酸治疗吉西他滨基础治疗后转移性胰腺癌(NAPOLI-1):一项全球、随机、开放标签的3期试验。《柳叶刀》387: 545 - 557。[Crossref]
- Reni M, Cereda S, Milella M, Novarino A, Passardi A,等(2013)转移性胰腺腺癌维持舒尼替尼或观察:一项II期随机试验。Eur J癌49: 3609 - 3615。[Crossref]
- Birk D, Beger HG(2001)胰腺癌的新辅助、辅助和姑息性治疗。柯尔消化科医生3: 129 - 135。[Crossref]
- De La Cruz MS, Young AP2, Ruffin MT2(2014)胰腺癌的诊断和治疗。Am Fam医生89: 626 - 632。(Crossref)
- Sikkens EC, Cahen DL, Kuipers EJ, Bruno MJ(2010)慢性胰腺炎胰酶替代治疗。最佳实践Res clinin肠胃醇24: 337 - 347。(Crossref)
- Costamagna G, Alevras P, Palladino F, Rainoldi F, Mutignani M,等(1999)内镜下胰腺支架在胰腺癌中的应用。J肠胃醇可以吗13: 481 - 487。(Crossref)
- 胰导管腺癌的内镜和手术姑息策略。Semin。肿瘤学杂志。42,163-176(2015)。
- 钟伟,于卓,曾建新,林燕,于涛,等。(2014)腹腔丛阻滞治疗胰腺癌相关疼痛的meta分析。疼痛Pract14: 43-51。(Crossref)
- Lili LN, Matyunina LV, Walker LD, Daneker GW, McDonald JF(2014)胰腺癌个人化分子谱分析重要性的证据。胰腺43: 198 - 211。[Crossref]
- 在胰腺癌的遗传进化过程中,远处转移发生较晚。自然467: 1114 - 1117。
- Sheikh R, Walsh N, Clynes M, O'Connor R, McDermott R(2010)胰腺癌治疗中的耐药性挑战。抗癌专家10: 1647 - 1661。(Crossref)
- Koay EJ1, Baio FE, Ondari A, Truty MJ, Cristini V,等。(2014)吉西他滨在人胰腺癌肿瘤内的传递和质粒运输异质性。期刊杂志11:0650002。[Crossref]
- 核糖核酸还原酶M1亚基过表达参与人胰腺癌吉西他滨耐药。Int。j .癌症120: 1355 - 1363(Crossref)
- Duxbury, m.s., Ito, H, Zinner, m.j., Ashley, S.W, Whang, E.E.(2004)靶向核糖核酸还原酶M2亚基的RNA干扰增强了胰腺癌患者对吉西他滨的化疗敏感性。致癌基因.23日:1539 - 1548(Crossref)
- Galmarini CM, Clarke ML, Falette N, Puisieux A, Mackey JR,等(2002)一种非功能性p53的表达影响癌细胞对吉西他滨的敏感性。Int J癌症97: 439 - 445。(Crossref)
- Bold, RJ, Chandra J, McConkey DJ(1999)吉西他滨诱导的人胰腺癌程序性细胞死亡(凋亡)由Bcl 2含量决定。安外科医生6: 279 - 285(Crossref)
- Ali(2010)通过姜黄素或其类似物CDF调节miR-200和miR-21的表达,可诱导胰腺癌细胞中吉西他滨的敏感性。癌症Res70: 3606 - 3617(Crossref)
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE(2004)抑制SRC酪氨酸激酶损害人胰腺癌细胞固有和获得性吉西他滨耐药。中国癌症Res10: 2307 - 2318(Crossref)
- NF kB和Akt/PI3K在胰腺癌细胞系抵抗吉西他滨诱导的细胞死亡中的作用。致癌基因22日:3243 - 3251(Crossref)
- Kasuya K(2011)胰腺癌低氧诱导因子1a表达与吉西他滨化疗。肿瘤防治杂志代表26日:1399 - 1406(Crossref)
- 吉西他滨耐药胰腺癌细胞上皮-间充质转变表型的获得与notch信号通路的激活有关。癌症Res69: 2400 - 2407。(Crossref)
- Ischenko I(2008)抑制Src酪氨酸激酶逆转人胰腺癌细胞对5-氟尿嘧啶的化疗耐药:与表皮生长因子受体信号通路有关。致癌基因27日:7212 - 7222(Crossref)
- 石霞,刘松,Kleeff J, Friess H, Büchler MW(2002)胰腺癌细胞对5-氟尿嘧啶和吉西他滨的获得性耐药与凋亡调节基因的表达改变有关。肿瘤学62: 354 - 362。[Crossref]
- Hagmann W, Jesnowski R, Faissner R, Guo C, Löhr JM(2009)人胰腺癌细胞系中atp结合盒C转运蛋白。5-氟尿嘧啶耐药细胞表达上调。胰脏学。9,136-144。[Crossref]
- Nambaru PK, Hübner T, Köck K, Mews S, Grube M,等(2010)药物外排转运体多药耐药相关蛋白5影响胰腺癌细胞系对核苷抗癌药物5-氟尿嘧啶的敏感性。药物Metab处理39: 132 - 139。[Crossref]
- Rachakonda PS, Bauer AS, Xie H, Campa D, Rizzato C,等(2013)外分泌胰腺肿瘤的体细胞突变:与患者生存的关系。《公共科学图书馆•综合》8: e60870。[Crossref]
- Cohen SJ (2003) farnesyltransferase抑制剂R115777作为转移性胰腺腺癌患者初始治疗的II期和药效学研究.J clinin Oncol2: 1301 - 6。
- Van Cutsem E, Van de Velde H, Karasek P, Oettle H, Vervenne WL等(2004)晚期胰腺癌吉西他滨加替法尼与吉西他滨加安慰剂的III期试验比较。J clinin Oncol22日:1430 - 1438。[Crossref]
- Lerner EC, Zhang TT, Knowles DB, Qian Y, Hamilton AD等(1997)抑制K-Ras的前酰化,但不抑制H-或N-Ras的前酰化,对CAAX类肽药物具有高度耐药性,需要法尼基转移酶和香叶酰香叶酰转移酶I抑制剂。致癌基因.15日,1283 - 1288。[Crossref]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q,等(2005)KRAS突变与肺腺癌对吉非替尼或厄洛替尼的原发性耐药。科学硕士2: e17。(Crossref)
- Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D等(2008)晚期结直肠癌中K-ras突变和西妥昔单抗的疗效。N英语J医学359: 1757 - 1765。[Crossref]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et . (2007) MET扩增通过激活ERBB3信号导致肺癌患者吉非他尼耐药。科学316: 1039 - 1043。[Crossref]
- Yamasaki F, Johansen MJ, Zhang D, Krishnamurthy S, Felix E,等(2007)A-431表皮样癌细胞获得性耐厄洛替尼需要下调MMAC1/PTEN和上调磷酸化Akt。Cancer Res. 67, 5779-5788。[Crossref]
- 陈海霞,Sharon E (2013) IGF-1R作为抗癌靶点的试验与磨难。中国癌症杂志32: 242 - 252。(Crossref)
- Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E.(2013)胰腺癌细胞中Akt和ERK反馈激活对雷帕霉素、活性位点mTOR抑制剂和二甲双胍的不同响应模式。《公共科学图书馆•综合》8: e57289。[Crossref]
- Michl P, Gress TM(2012)改善胰腺癌药物输送:通过靶向透明质酸打破基质堡垒。肠道6:1377 - 1379。[Crossref]
- Olive KP1, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D,等(2009)抑制hedgehog信号通路增强胰腺癌小鼠模型化疗的传递。科学324: 1457 - 1461。[Crossref]
- Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK,等(2014)hedgehog途径抑制剂GDC-0449 (vismodegib)联合吉西他滨治疗转移性胰腺腺癌的临床初步研究。临床癌症治疗20: 5937 - 5945。[Crossref]
- De Jesus-Acosta A(2014)一项关于hedgehog (Hh)途径抑制剂vismodegib联合吉西他滨和nab-紫杉醇(nabp)治疗未治疗的转移性胰腺导管腺癌(PDA)患者的II期研究。J clinin Oncol32(增刊):257。
- Catenacci, D.V.T.等(2015)吉西他滨联合安慰剂或hedgehog途径抑制剂vismodegib在转移性胰腺癌患者中的随机Ib/ II期研究。J clinin Oncol33: 4284 - 4292。[Crossref]
- Lee JJ, Perera RM, Wang H, Wu DC, Liu XS,等(2014)Hedgehog信号通路的基质反应抑制胰腺癌进展。美国国家科学研究院11: 3091 - 3100。[Crossref]
- Grützmann R, Boriss H, Ammerpohl O, Lüttges J, Kalthoff H,等(2005)胰腺癌微阵列数据的meta分析定义了一组常见的失调基因。致癌基因24: 5079 - 5088。(Crossref)
- Miyamoto H, Murakami T, Tsuchida K, Sugino H, Miyake H,等(2004)人胰腺癌肿瘤-间质相互作用:获得性耐药和增殖调节依赖于细胞外基质蛋白。胰腺28: 38-44。[Crossref]
- Toste PA, Nguyen AH, Kadera BE, Duong M, Wu N,等(2016)通过应激相关的MAPK在胰腺CAFs中化疗诱导的炎症基因特征和致瘤表型。Mol Cancer Res[Crossref]
- Morimoto M, Matsuo Y2, Koide S, Tsuboi K, Shamoto T,等(2016)胰腺癌患者获得吉西他滨耐药导致CXCL12/CXCR4轴增强:CXCR4拮抗剂的作用。BMC癌症16: 305。(Crossref)
- Royal RE, Levy C, Turner K, Mathur A, Hughes M等人(2010)单药伊匹单抗(抗ctla -4)治疗局部晚期或转移性胰腺腺癌的2期试验。J Immunother33: 828 - 833。[Crossref]
- Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC,等。(2016)抑制肝细胞生长因子:一种新的胰腺癌治疗方法。BJ癌症114:269 - 280。[Crossref]
- 鲍森,吴强,McLendon RE,郝毅,石青,等(2006)胶质瘤干细胞通过优先激活DNA损伤反应促进辐射抗性。自然444: 756 - 760。[Crossref]
- Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS,等(2004)造血干细胞增殖和静止的分子特征。公共科学图书馆杂志2: e301。(Crossref)
- Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W,等(2011)在人胰腺癌中,ALDH活性选择性地定义了相对于CD133表达增强的肿瘤起始细胞群。《公共科学图书馆•综合》6: e20636。[Crossref]
- O 'Reilly EM (2015) brca突变胰腺腺癌:新出现的治疗意义。AACR胰腺癌特别会议。会议摘要癌症Res75.
- O 'Reilly EM(2014)顺铂、吉西他滨和威利帕尼在已知或潜在BRCA或PALB2突变胰腺腺癌患者中的IB期试验。J clinin Oncol为了。
- Venkatasubbarao K, Peterson L, Zhao S, Hill P, Cao L,等(2013)抑制转录-3信号转导和激活因子增加对吉西他滨的反应并延缓胰腺癌进展。摩尔癌症12:104。[Crossref]
- Slusarczyk M, Lopez MH, Balzarini J, Mason M, Jiang WG, et al. (2014) ProTide技术在吉西他滨上的应用:一种成功克服关键癌症抵抗机制的方法导致一种新的药物(nuci -1)在临床开发中。医学化学57: 1531 - 1542。[Crossref]
- Blagden S (2015) nuu -1031在实体肿瘤患者中的首次人类I/II期研究的最终结果。ASCO。摘要号:2514。
- Gilabert M, Calvo E, Airoldi A, Hamidi T, Moutardier V等(2014)小鼠胰腺癌诱导的恶病质依赖于jak2。细胞物理229: 1437 - 1443。(Crossref)
- Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT等(2015)Ruxolitinib或安慰剂联合卡培他滨对吉西他滨治疗失败的转移性胰腺癌患者的随机、双盲II期研究。J clinin Oncol33: 4039 - 4047。(Crossref)
- 李海生,李盛杰,李海杰,Chung MJ, Park JY,等(2016)胰腺癌患者使用他汀类药物及其对生存的影响。医学(巴尔的摩)95: e3607。(Crossref)
- Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D(2012)糖尿病合并胰腺癌患者使用二甲双胍与更好的生存期相关。临床癌症治疗18: 2905 - 2912。[Crossref]
- 张鹏,李慧,谭旭,陈磊,王松(2013)二甲双胍使用与癌症发病率和死亡率的相关性:一项meta分析。癌症的论文37: 207 - 218。(Crossref)
- Fasih A, Elbaz HA, Hüttemann M, Konski AA, Zielske SP(2014)二甲双胍通过AMPK途径对胰腺癌细胞的放射增敏作用。Radiat Res50 182: 59。[Crossref]
- Karnevi E, Said K, Andersson R, Rosendahl AH(2012)二甲福明介导的生长抑制涉及抑制人胰腺癌细胞中IGF-I受体信号通路。BMC癌症13: 235。[Crossref]
- Wilmink J(2014)一项II期随机、安慰剂对照研究评估吉西他滨、厄洛替尼和二甲双胍联合治疗局部晚期或转移性胰腺癌的疗效。ASCO年会。J clinin Oncol.芝加哥。
- Braghiroli MI, de Celis Ferrari AC, Pfiffer TE, Alex AK, Nebuloni D等(2015)二甲双胍和紫杉醇联合治疗吉西他滨难治性晚期胰腺腺癌的II期临床研究。Ecancer9: 563。[Crossref]
- Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, de Oliveira E,等(2013)Notch信号通路靶向治疗抑制胰腺癌肿瘤进展和转移扩散。癌症列托人335: 41-51。(Crossref)
- Mueller MT(2009)联合靶向治疗消除人胰腺癌中的致瘤性癌症干细胞。胃肠病学137: 1102 - 1113。[Crossref]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, et . (2015) MYC/PGC-1α平衡决定胰腺癌干细胞的代谢表型和可塑性。细胞金属底座22日:590 - 605。(Crossref)
- 赵L,赵Y,施瓦兹B, Mysliwietz J, Hartig R, et al。(2016)异搏定抑制肿瘤进展chemotherapy-resistant胰腺癌一边人口的细胞。Int J Oncol49: 99 - 110。[Crossref]
- Yardley DA (2013) nabe -紫杉醇的作用和传递机制。J控制释放170: 365 - 372。(Crossref)
- Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS等(2011)吉西他滨+ nab-紫杉醇是晚期胰腺癌患者的有效方案:I/II期试验。J clinin Oncol29日:4548 - 4554。(Crossref)
- Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D,等。(2013)纳米粒-紫杉醇在胰腺癌中的基质干扰作用。乳腺癌109: 926 - 933。(Crossref)
- Gonzalez-Villasana V, Rodriguez-Aguayo C, Arumugam T, Cruz-Monserrate Z, Fuentes-Mattei E等(2014)双膦酸盐抑制胰导管腺癌星形细胞活性并增强纳米颗粒白蛋白结合紫杉醇的抗肿瘤作用。其他癌症13: 2583 - 2594。[Crossref]
- Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC,等。(2016)抑制肝细胞生长因子:一种新的胰腺癌治疗方法。乳腺癌114: 269 - 280。[Crossref]
- Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC,等(2016)Hif1a缺失揭示了B细胞在胰腺肿瘤中的促肿瘤功能。癌症越是加大6: 256 - 269。[Crossref]
- Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI,等(2016)布鲁顿酪氨酸激酶(BTK)依赖性免疫细胞串声驱动胰腺癌。癌症越是加大6: 270 - 285。[Crossref]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR,等(2011)CD40激动剂改变了肿瘤间质,并对小鼠和人的胰腺癌显示出疗效。科学33: 1612 - 1616。[Crossref]
- Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A等(2013)一项激动剂CD40单克隆抗体(CP-870,893)联合吉西他滨治疗晚期胰导管腺癌的I期研究。临床癌症治疗19日:6286 - 6295。[Crossref]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A,等(2013)靶向表达fap的癌相关成纤维细胞CXCL12与抗pd - l1免疫治疗在胰腺癌中的协同作用。美国国家科学研究院110: 20212 - 20217。[Crossref]
- Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S,等(2012)用于晚期癌症的李斯特菌减毒活疫苗(ANZ-100)和表达间皮素的李斯特菌减毒活疫苗(CRS-207):安全性和免疫诱导的I期研究。临床癌症治疗18: 858 - 868。[Crossref]
- Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T,等(2015)GVAX胰源和表达单核增生李斯特菌间皮素(CRS-207)增强疫苗治疗转移性胰腺癌的安全性和生存率。J clinin Oncol33:1325 - 1333。[Crossref]
- McEwan C, Owen J, Stride E, Fowley C, Nesbitt H,等。(2015)载氧微泡增强声动力治疗低氧肿瘤。J控制释放203: 51-56。[Crossref]
- McEwan C, Kamila S, Owen J, Nesbitt H, Callan B,等。结合声学和抗代谢药物治疗,以载氧微泡作为运载工具改善胰腺癌的治疗。生物材料80: 20-32(2016)。[Crossref]
- Mullard A(2014)向卓越的药物反应者学习。Nat Rev药物发现13: 401 - 402。(Crossref)
- Iyer G, Hanrahan AJ, Milowsky MI, al - ahmadie H, Scott SN,等(2012)基因组测序确定依维莫司敏感性的基础。科学338: 221。[Crossref]
- Subbiah V, Naing A, Brown RE, Chen H, Doyle L,等。(2011)胰岛素样生长因子1受体(IGF1R)抑制剂治疗尤文氏肉瘤的靶向形态蛋白质组学分析:反应/抵抗特征。《公共科学图书馆•综合》6: e18424。[Crossref]
- Chue BM(2009)转移性胰腺癌5年生存率:勇气和希望的研究。胃肠癌症保留区3: 208 - 211。(Crossref)
- Klimant E, Markman M, Albu DM(2013)转移性胰腺癌患者序贯吉西他滨化疗方案的临床意义应答。案件代表6: 72 - 77。[Crossref]
- Rios Perez MV, Dai B, Koay EJ, Wolff RA, Fleming JB (2015) IV期胰腺癌对治疗性手术的回归和一种新的体外化疗敏感性测定方法的引入。Cureus7: e423。[Crossref]
- 岑鹏,倪旭,杨杰,Graham DY,李明(2012)循环肿瘤细胞在胰腺癌诊断和治疗中的作用。生物化学生物生物学学报1826: 350 - 356。[Crossref]
- Poruk KE, Valero V 3rd, Saunders T, Blackford AL, Griffin JF等(2016)循环肿瘤细胞表型预测胰腺腺癌的复发和生存。安杂志264:1073 - 1081。[Crossref]
- Garcea G, Ladwa N, Neal CP, Metcalfe MS, Dennison AR,等(2011)术前中性粒细胞与淋巴细胞比值(NLR)与胰腺腺癌根治性切除术后无病生存期降低相关。世界J外科35: 868 - 872。[Crossref]
- Radon TP, Massat NJ, Jones R, Alrawashdeh W, dummartin L,等。(2015)尿液中三生物标志物的鉴定用于胰腺腺癌的早期检测。临床癌症治疗2: 3512。[Crossref]
- 大坪K, Watanabe H, Yao F, Okada G, Mouri H,等。纯胰液中前脑啡肽高甲基化与p53突变在胰腺癌诊断中的比较。杂志4: 791-797(2006)。[Crossref]
- Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA,等(2010)6个基因特征预测局限性胰腺导管腺癌患者的生存。科学硕士7: e1000307。(Crossref)
- Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S,等(2010)细胞组蛋白修饰模式预测可切除胰腺腺癌的预后和治疗反应:RTOG 9704的结果。J clinin Oncol28日:1358 - 1365。[Crossref]
- Moravec R, Divi R, Verma M(2017)胰腺癌进展过程中循环肿瘤物质的检测和数字病理成像。世界胃肠病学9: 235 - 250。[Crossref]
- Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST等(2015)Glypican1可识别癌症外泌体并促进癌症早期检测。自然杂志523:177-182。[Crossref]
- Ghazaly EA(2015)验证RNAscope用于吉西他滨耐药相关关键生物标志物的分子分析。癌症Res75: 3390。
- Orlandi A, Calegari MA, Martini M, Cocomazzi A, Bagalà C,等人(2016)吉西他滨与FOLFIRINOX在晚期hent1阳性胰腺腺癌患者中的疗效比较:当一切似乎更糟时,一切都不太糟。clinin Transl Oncol18: 988 - 995。[Crossref]
- Hudis CA(2007)曲妥珠单抗的作用机制和临床应用。N英语J医学357: 39-51。(Crossref)
- 王金平,吴春春,叶永春,薛玉梅,吴云云,等(2015)厄洛替尼对胰腺癌表皮生长因子受体突变有效:一项随机、开放标签的前瞻性试验。Oncotarget6: 18162 - 73。[Crossref]
- Golan T, Atias D, Barshack I, Avivi C, Goldstein RS,等(2014)腹水源性胰导管腺癌原代细胞培养作为个体化药物平台。乳腺癌110: 2269 - 2276。[Crossref]
- 张洪昌,郭cj(2015)利用hPSCs对胰腺癌类器官进行个体化研究。Nat地中海21日:1249 - 1251。(Crossref)
- Keshet Y, Schiff E, Samuels N, Ben-Arye E(2015)为癌症患者发声:通过简短的患者叙事评估综合肿瘤治疗方案的非特异性效果。Psychooncology24: 169 - 174。[Crossref]
- Sagar SM, Leis AM(2008)综合肿瘤学:加拿大和国际视角。咕咕叫杂志15补充2:s71-73。(Crossref)
- 邓G, Cassileth B(2013)癌症治疗中的补充或替代医学-神话和现实。Nat Rev clinin Oncol10: 656 - 664。(Crossref)
- Marchand L(2014)晚期癌症患者的综合和补充疗法。安·帕里特医生3: 160 - 171。(Crossref)
- Campbell KL, McTiernan A(2007)运动和癌症预防研究中的生物标志物。J减轻137: 161 - 169年代。(Crossref)
- 世界癌症研究基金/美国癌症研究所(2007)食物营养、身体活动和癌症预防:全球视角。华盛顿特区:美国癌症研究所。
- Grivennikov SI, Greten FR, Karin M(2010)免疫、炎症和癌症。细胞140: 883 - 899。(Crossref)
- Neuzillet C, Vergnault M, Bonnetain F, Hammel P(2015)晚期胰腺癌患者适应性体育活动(APACaP)的理论基础和设计GERCOR (Groupe Coopérateur多学科肿瘤学)试验:随机对照试验的研究方案。试用16: 454。[Crossref]
- Frenkel M, Sierpina V, Sapire K(2015)补充和结合医学对癌症生存率的影响。Curr Oncol代表17: 445。(Crossref)
- Sagar SM, Lawenda BD(2009)综合肿瘤学在三级预防生存计划中的作用。Prev地中海49: 93 - 98。[Crossref]
- Yang EV, Glaser R(2003)应激诱导的免疫调节:对肿瘤发生的影响。大脑行为17: 37-S40。[Crossref]
- Chan C, Lin HJ, Lin J(2008)应激相关激素去甲肾上腺素可增加人胰管上皮细胞的增殖和IL-6水平,并可被膳食制剂萝卜硫素抑制。Int J Oncol33: 415 - 419。[Crossref]
- Genkinger JM, Wang M, Li R, Albanes D, Anderson KE,等(2014)乳制品与胰腺癌风险:14项队列研究的汇总分析。安杂志25:1106 - 1115。[Crossref]
- Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN(2010)卡路里与致癌:从30年卡路里限制研究中吸取的教训。致癌作用3: 83 - 89。[Crossref]
- Lashinger LM, Malone LM, Brown GW, Daniels EA, Goldberg JA, et al.(2011)雷帕霉素在胰腺癌小鼠模型中部分模仿卡路里限制的抗癌效果。癌症预防(Phila)4: 1041 - 51。[Crossref]
- Harvey AE, Lashinger LM, Hays D, Harrison LM, Lewis K,等(2014)卡路里限制以胰岛素样生长因子-12依赖的方式降低小鼠和人类胰腺肿瘤细胞生长、核因子- kb激活和炎症相关基因表达。《公共科学图书馆•综合》9: E94151。[Crossref]
- Lanza-Jacoby S, Yan G, Radice G, LePhong C, Baliff J, et al.(2013)卡路里限制延缓LSL-KrasG12D中胰腺癌病变的进展;Pdx-1/Cre小鼠胰腺癌模型。高级生物医生(梅坞)238: 787 - 97。[Crossref]
- Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V等(2014)酮体诱导的代谢重编程减少胰腺癌恶病质。癌症金属底座18。[Crossref]
- Uomo G, Gallucci F, Rabitti PG(2006)胰腺癌的厌食-恶病质综合征:研究和管理的最新进展。事的话7: 157 - 62。[Crossref]
- Hashim SA, VanItallie TB1(2014)酮体疗法:从生酮饮食到口服酮酯。J脂质Res55岁:1818 - 1826。(Crossref)
- Tang SN, Fu J, Shankar S, Srivastava RK (2012) EGCG通过抑制STAT3信号通路增强吉西他滨和CP690550在人胰腺癌中的治疗潜力。《公共科学图书馆•综合》7: e31067。[Crossref]
- Kanai M, Yoshimura K, Asada M, Imaizumi A, Suzuki C,等。(2011)吉西他滨耐药胰腺癌患者吉西他滨化疗加姜黄素的I/II期研究。癌症化学,其他药典68: 157 - 164。[Crossref]
- Li Y, VandenBoom TG 2nd, Kong D, Wang Z, Ali S, et al.(2009)自然因子上调miR-200和let-7导致吉西他滨耐药胰腺癌细胞上皮向间质转变逆转。癌症Res69: 6704 - 6712。[Crossref]
- Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, et al.(2010)白藜芦醇是一种多靶点制剂,可增强吉西他滨在体外和原位小鼠胰腺癌模型中的抗肿瘤活性。Int J癌症127: 257 - 268。[Crossref]
- Arshad A, Isherwood J, Mann C, Cooke J, Pollard C,等(2015)静脉注射ɷ-3脂肪酸加吉西他滨:改善晚期胰腺癌反应和生活质量的潜力。父母肠内坚果[Crossref]
- Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D,等(2010)胰腺癌切除术后氟脲嘧啶+叶酸辅助化疗vs吉西他滨:一项随机对照试验。《美国医学会杂志》304: 1073 - 1081。[Crossref]
- Cheng X, Kim JY, Ghafoory S, Duvaci T, Rafiee R, et al.(2016)甲基异蓝靛通过抑制pdac中LKB1和激活AMPK干扰细胞代谢,优先杀死癌症干细胞。摩尔杂志10: 806 - 824。[Crossref]
- 张颖,陈爱英,李明,陈超,姚强(2008)银杏叶提取物山奈酚抑制胰腺癌细胞增殖并诱导凋亡。外科手术148: 17-23。[Crossref]
- Ferguson LR, Chen H, Collins AR, Connell M, Damia G,等(2015)人类癌症的基因组不稳定性:通过饮食和营养的分子洞察和治疗攻击和预防的机会。Semin Cancer Biol35岁,5-24。[Crossref]
- Banerjee S,张颖,王铮,车敏,焦丕杰,等(2006)染料木素增强顺铂治疗胰腺癌疗效的体内外分子证据。Int J癌症120: 906 - 917。[Crossref]
- Bhattacharjee Y, Zhou Y, Yen TJ(2014)一种合成致死筛选识别了胰腺癌细胞中维生素D受体作为一种新型吉西他滨致敏剂。细胞周期13: 3838 - 3856。[Crossref]
- Persons KS, Eddy VJ, Chadid S, Deoliveira R, Saha AK,等。(2010)25-二羟基维生素d3 -3-溴乙酸酯单独或联合5-氨基咪唑-4-羧酰胺-1- β -4-核呋喃苷对胰腺癌细胞生长的抑制作用。抗癌物30: 1875 - 1880。[Crossref]
- Penberthy DR, Rich TA, Shelton CH 3rd, Adams R, Minasi JS等(2001)胰腺癌定时输液5-氟尿嘧啶放化疗的初步研究。安杂志12: 681 - 4。[Crossref]
- Relles D, Sendecki J, Chipitsyna G, Hyslop T, Yeo CJ,等。(2013)胰腺癌昼夜基因表达与临床病理的相关性。胃肠外科杂志17: 443 - 450。(Crossref)
- Oda A, Katayose Y, Yabuuchi S, Yamamoto K, Mizuma M, et .(2009)时钟基因小鼠period2过表达抑制人胰腺癌细胞生长并与顺铂具有协同作用。抗癌物29日:1201 - 1209。[Crossref]
- 李晓明(2010)通过饮食时间对肿瘤转录组的昼夜重编程抑制癌症。癌症Res70: 3351 - 60。[Crossref]
- Balasenthil S, Chen N, Lott ST, Chen J, Carter J,等。(2011)胰腺腺癌的迁移特征和血浆生物标志物面板。Cancer Res (Phila)4:137 - 149。[Crossref]
- Brand RE, Nolen BM, Zeh HJ, Allen PJ, Eloubeidi MA,等(2011)用于胰腺癌检测的血清生物标志物面板。临床癌症治疗17: 805 - 816。(Crossref)
- Radon TP, Massat NJ, Jones R, Alrawashdeh W, dummartin L,等。(2015)尿液中三生物标志物的鉴定用于胰腺腺癌的早期检测。临床癌症治疗2: 3512。[Crossref]
- 王杰,陈杰,常萍,LeBlanc A,李迪等(2009)胰腺导管腺癌患者血浆中的MicroRNAs作为一种新的基于血液的疾病生物标志物。癌症:Res(费拉)2:807 - 813。[Crossref]
- Pedersen KS, Bamlet WR, Oberg AL, de Andrade M, Matsumoto ME, et AL .(2011)白细胞DNA甲基化特征将胰腺癌患者与健康对照组区分开来。《公共科学图书馆•综合》6: e18223。[Crossref]
- Wingren C, Sandström A, Segersvärd R, Carlsson A, Andersson R,等(2012)胰腺癌相关血清生物标志物的鉴定。癌症Res72: 2481 - 2490。(Crossref)
- Bracci PM, Zhou M, Young S, Wiemels J(2012)一项旧金山湾区病例对照研究中胰腺癌抗原血清自身抗体作为胰腺癌的生物标志物。癌症118: 5384 - 5394。[Crossref]
- Tanaka M, Javle M, Dong X, Eng C, Abbruzzese JL,等(2010)局部晚期胰腺癌患者吉西他滨代谢和转运蛋白基因多态性与药物毒性和疗效相关。癌症116: 5325 - 5335。[Crossref]
- Woo HI, Kim KK, Choi H, Kim S, Jang KT,等(2012)基因多态性对吉西他滨治疗胰腺癌患者疗效和临床结局的影响。药物基因组学13: 1023 - 1035。[Crossref]
- Lemstrová R1, souek P, Melichar B, Mohelnikova-Duchonova B(2014)溶质载体转运蛋白在胰腺癌中的作用:综述。药物基因组学15:1133 - 1145。[Crossref]
- Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T,等(2010)基因表达水平作为胰腺癌吉米他滨化疗后预后的预测指标。瘤形成12: 807 - 817。[Crossref]
- Tang SN, Fu J, Shankar S, Srivastava RK (2012) EGCG通过抑制STAT3信号通路增强吉西他滨和CP690550在人胰腺癌中的治疗潜力。《公共科学图书馆•综合》7: e31067。[Crossref]
- Li L, Leung PS(2014)使用草药和天然产物:一种克服胰腺癌凋亡耐药性的替代方法。生物化学细胞生物学杂志53: 224 - 236。[Crossref]
- Boreddy SR, Srivastava SK2(2013)植物化学物质对胰腺癌的化学预防。癌症列托人334: 86 - 94。(Crossref)
- Gong Z, Holly EA, Bracci PM(2009)叶酸、维生素B6、B12和蛋氨酸摄入量与胰腺癌风险的一项大型人群病例对照研究。癌症成因控制20: 1317 - 1325。[Crossref]
- Donahue TR, Tran LM, Hill R, Li Y, Kovochich A,等(2012)人胰腺癌综合生存分子谱分析。临床癌症治疗18: 1352 - 1363。(Crossref)
- Nakahara O, Takamori H, Tanaka H, Sakamoto Y, Ikuta Y,等。(2010)胰腺癌患者二氢嘧啶脱氢酶和胸苷酸合成酶表达的临床意义。Int J clinin Oncol15: 39-45。[Crossref]
- Neoptolemos JP(2009)辅助5-氟尿嘧啶和叶酸对胰腺癌的观察:ESPAC-1和-3(v1)试验的综合数据。乳腺癌100: 246 - 250。
- Strippoli A (2016) ERCC1表达对FOLFIRINOX治疗转移性胰腺癌的疗效影响:单机构分析。Oncotarget7: 35159 - 35168。[Crossref]
- 李东,刘红,焦琳,常德志,Beinart G,等(2006)同源重组DNA修复基因多态性对胰腺癌生存的显著影响.癌症Res66: 3323 - 3330。[Crossref]
- Boeck S, Jung A, Laubender RP, Neumann J, Egg R,等(2013)厄洛替尼治疗的晚期胰腺癌患者中的EGFR通路生物标志物:来自随机、交叉三期临床试验AIO-PK0104的翻译结果。乳腺癌108: 469 - 476。[Crossref]
- Vaz J, Ansari D, Sasor A, Andersson R (2015) SPARC:胰腺癌潜在的预后和治疗靶点。胰腺44: 1024 - 1035。(Crossref)





